
The Kelly Group Research
Gelsolin amyloid disease mechanism and therapeutics:
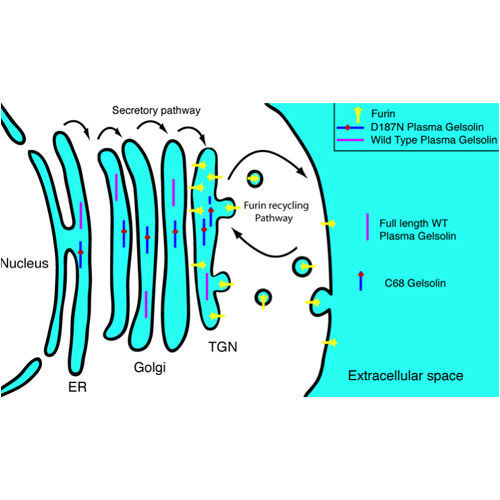
Figure 1: Schematic representation of plasma gelsolin secretion (Page et al., Ca2+binding protects against gelsolin amyloidosis. Biochem. Biophys. Res. Comm. 2004;322:1105-1110). Full-length wild-type and D187N/Y mutant gelsolin are synthesized in the endoplasmic reticulum (ER). The trans-Golgi network (TG), endoplasmic reticulum and serum contain high Ca2+ concentrations (blue) which protects wild-type gelsolin against proteolytic cleavage by the α-gelsolinase furin. The D187N/Y mutant gelsolin does not bind Ca2+ in domain 2 and is consequently cleaved by furin generating a C-terminal 68 kDa fragment. The latter is secreted into the extracellular matrix and further cleaved by β-gelsolinase, yielding the 8 and 5 kDa amyloidogenic peptides characteristically present in patients with hereditary gelsolin amyloidosis. Courtesy of Dr. Lesley Page.
Hereditary gelsolin amyloidosis is caused by the D187N/Y mutation in the actin manipulating protein, plasma gelsolin, that leads to amyloid deposits comprised of 8 kDa (major component) and 5 kDa (minor component) internal gelsolin fragments. We discovered that the aberrant proteolytic step triggering gelsolin amyloidosis is caused by the loss of Ca2+ binding in domain 2, which destabilizes the protein as it traffics through the trans-Golgi. This domain 2 destabilization allows a fraction of the mutant gelsolin protein to be cut by the protease furin, leading to the secretion of a 68 kDa fragment. In contrast, wild-type plasma gelsolin is not susceptible to cleavage by furin and is secreted as a full length nonamyloidogenic protein. The 68 kDa fragment undergoes further cleavage by β-gelsolinase, resulting in the 8 and 5 kDa fragments that deposit as amyloid. We have identified MT1-MMP as one β-gelsolinase candidate using a cell line and are actively engaged in exploring the identity(ies) of β-gelsolinase in mice and human patients. A subset of the glycosaminoglycans present in the extracellular matrix accelerates gelsolin amyloidogenesis, possibly explaining the tissue specificity of this disease.
In collaboration with the laboratory of Dr. Balch, we have recently developed two transgenic mouse lines which recapitulate many aspects of the human disease. We anticipate that these mouse models will be critically important for developing gelsolin amyloid disease therapeutics. The mice generate the 68, 8 and 5 kDa proteolytic fragments in muscle; they deposit 8 kDa, and to a lesser degree, 5 kDa fragments in the glycosaminoglycan-rich region surrounding the muscle cells; only the 68 kDa fragment and full length gelsolin is found in the blood; muscle pathology is observed late in the disease; and muscle weakness is measurable in the D187N line expressing higher levels of the transgene.
Only about 20-40% of the mutated protein is aberrantly cleaved by furin within the secretory pathway, ultimately affording amyloid and causing disease. Thus, if it were possible to decrease the amount of misfolded protein or reduce the concentration of the amyloidogenic fragments by only a small amount, symptoms may be alleviated because the process of amyloidogenesis is very dependent on precursor concentration. We are currently exploring two possible strategies for the amelioration of gelsolin amyloidosis: 1) proteostasis regulators, small molecules that restore normal protein homeostasis, to improve the folding and trafficking of mutant gelsolin, and 2) β-gelsolinase inhibitors to prevent the formation of the 8 kDa amyloidogenic fragment.
We also seek to better understand the mechanism of gelsolin amyloidogenesis and to identify the structures associated with toxicity, focusing on the role of glycosaminoglycans in the tissue specificity of these diseases. We have shown that the 8 and 5 kDa gelsolin fragments aggregate via a nucleated polymerization mechanism and that the 68 kDa fragment cannot form fibrils, although it can reversibly oligomerize. We are currently further investigating how glycoaminoglycans influence gelsolin amyloidogenesis.
- Page LJ, Huff ME, Kelly JW, Balch WE. Ca2+ binding protects against gelsolin amyloidosis. Biochem. Biophys. Res. Comm.2004; 322:1105-1110.
- Page LJ, Suk J Y, Huff ME, Lim H-J, Venable J, Yates III J, Kelly JW, Balch WE. Metalloendoprotease cleavage triggers gelsolin amyloidogenesis. EMBO J. 2005; 24:4124-4132.
- Suk JY, Zhang F, Balch WE, Linhardt R, Kelly JW. Heparin accelerates gelsolin amyloidogenesis. Biochemistry 2006;45:2234-2242.
- Page LJ, Suk J-Y, Bazhenova L, Fleming SM, Wood M, Guo LT, Misizin AP, Kisilevsky R, Shelton DG, Balch WE, Kelly JW. Secretion of Amyloidogenic Gelsolin Progressively Compromises Protein Homeostasis Leading to the Intracellular Aggregation of Proteins. Proc. Natl. Acad. Sci.2009; 106:11125-11130.
-
- Solomon JP, Yonemoto IT, Murray AN, Price JL, Powers ET, Kelly JW. Plasma Gelsolin 5 and 8 kDa Fragments Form Amyloid by a Nucleated Polymerization Mechanism, while the 68 kDa Fragment is Not Amyloidogenic. Biochemistry2009;48:11370-11380.
Introduction
Transthyretin amyloid diseases: understanding the mechanism of proteotoxicity and inhibition of amyloid fibril formation
Understanding Abeta aggregation and Alzheimer's disease
Gelsolin amyloid disease mechanism and therapeutics
β-Sheet folding
Parkinson's disease
Restoration of enzyme homeostasis in lysosomal storage diseases
Amyloid as a functional fold
California: 10550 North Torrey Pines Road, La Jolla, CA 92037 - (858) 784-1000
Florida: 130 Scripps Way, Jupiter, FL 33458 - (561) 228-2000
Copyright © 2025, The Scripps Research Institute (TSRI). All Rights Reserved.