
The Kelly Group Research
Restoration of enzyme homeostasis in lysosomal storage diseases:
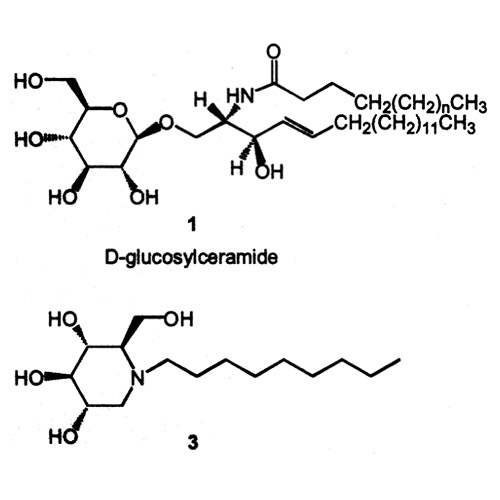
Figure 1: Mutations (e.g. N370S) in the gene encoding glucocerebrosidase, an enzyme that converts D-glucosylceramide into ceramide and D-glucose, lead to an accumulation of D-glucosylceramide (1). Addition of chemical chaperone N-(n-nonyl)-deoxynojirimycin (3) to N370S fibroblast cultures leads to a significant increase in enzyme activity (Sawkar et al. Chemical chaperones increase the cellular activity of N370S β-glucosidase: a therapeutic strategy for Gaucher disease. Proc. Natl. Acad. Sci. USA. 2002;99:15428-15433). Courtesy of Anu Sawkar.
Lysosomal storage diseases (LSDs) result from deficient lysosomal enzyme activity leading to accumulation of the substrate of the mutant lysosomal enzyme. At least 40 distinct LSDs have been identified with the most prevalent being Gaucher disease, which is caused by a deficiency in the activity of lysosomal glucocerebrosidase (GC). Gaucher disease affects 1 in 60,000 people in the general population and 1 in 800 among the Ashkenazi Jewish population. In many LSDs, mutations compromise the folding of the enzyme in the endoplasmic reticulum (ER) subjecting it to ER-associated degradation instead of proper folding and trafficking to the lysosome. Lowering the growth temperature of patient-derived fibroblasts dramatically improves mutant enzyme folding efficiency in the ER, enabling the mutant folded enzyme to engage its trafficking receptor and be trafficked to the lysosome where the enzyme is functional, even when the growth temperature is increased to 37 °C. We have and are currently investigating pharmacological approaches to improve the folding, trafficking and function of the mutant enzymes associated with LSDs.
In the first approach, we have shown that small molecules can act as pharmacologic chaperones, stabilizing the folded and near-folded forms of the enzymes in the ER, increasing the population of folded lysosomal enzymes that can engage their trafficking receptor in the ER and be trafficked to the lysosome. This enables more enzyme to be trafficked successfully from the ER to the lysosome, where it remains stable and functional in the absence of bound chaperone owing to the acidic environment in the lysosome in which these enzymes were evolved to function in. Thus, the small molecule N-(n-nonyl)-deoxynojirimycin (Figure 1) can increase the activity of N370S GC, the most common Gaucher disease-associated mutant, in patient-derived cells. Additional classes of small molecules (such as deoxynojirimycin analogues and isofagomines) are able to increase the activity of various GC mutants (such as N370S and G202R). The neuropathic GC mutant, L444P, is not amenable to chemical chaperoning because of the very low concentration of folded protein present in the ER and because this mutant enzyme is highly susceptible to inhibition. The drawback to this approach is that since each pharmacologic chaperone is enzyme specific, a different pharmacologic chaperone has to be created for each lysosomal enzyme associated with a LSD.
In the next two approaches, we aim to discover proteostasis regulators, small molecules that increase the capacity of the proteostasis network in the ER and thus enhance the folding, trafficking and function of lysosomal enzymes associated with multiple LSDs involving non-homologous enzymes. Proteostasis refers to controlling the concentration, conformation, binding interactions (quaternary structure), and location of individual proteins making up the proteome by readapting the innate biology of the cell, often through transcriptional and translational changes. Proteostasis is influenced by the chemistry of protein folding/misfolding and by numerous regulated networks of interacting and competing biological pathways comprising the proteostasis network. Proteostasis network components include the highly regulated ribosome that synthesizes proteins, the chaperone-chaperonin-enzyme-mediated folding networks, the trafficking components, disaggregation pathways, and proteasome- and autophagy-mediated degradation. First, we outline an approach where endoplasmic reticulum proteostasis is enhanced by a post-translational mechanism, and secondly, we outline an approach where lysosomal enzyme proteostasis is enhanced by a transcriptional mechanism.
Regarding post-translational regulation of endoplasmic reticulum proteostasis, we have shown that increasing ER calcium levels can enhance the folding, trafficking, and activity of mutant lysosomal enzymes in patient-derived fibroblasts. Partial restoration of activity was observed for both the N370S GC mutation and the more refractory, neuropathic L444P GC mutation. We used the FDA-approved drugs, diltiazem and verapamil, which inhibit both the L-type calcium channels and the ryanodine receptors. We have also specifically targeted the ryanodine receptors using dantrolene. Antagonism of the ryanodine receptors appears to be sufficient to increase ER calcium levels, which enhances the capacity of the calnexin chaperone system to fold mutant lysosomal enzymes in Gaucher disease, α-mannosidosis and type IIIA mucopolysaccharidosis. Thus, manipulation of calcium homeostasis may represent a general strategy to restore enzyme proteostasis in multiple LSDs.
In an alternative approach, we have shown that activation of the unfolded protein response (UPR) stress-responsive signaling pathway is effective at enhancing enzyme proteostasis in multiple LSDs. Activation of the UPR results in transcription and translational increases that enhance the capacity of the ER to fold and traffic proteins. Treatment with two distinct proteostasis regulators that activate the UPR, celastrol and MG-132, partially restores mutant enzyme folding, trafficking and function in several LSDs, including L444P GC activity in Gaucher patient-derived cell lines. This is exciting because the L444P GC mutation that often leads to neuropathic Gaucher disease does not respond significantly to pharmacologic chaperones for reasons described above. Currently more selective means of activating the UPR are being developed by our laboratory.
We have also shown that the combined use of a proteostasis regulator and an enzyme-specific pharmacologic chaperone synergistically restores mutant enzyme function in Gaucher and Tay-Sachs patient-derived cell lines, owing to their distinct mechanisms of action. Thus, another strategy to restore enzyme homeostasis may be through administration of proteostasis regulators in combination with a pharmacologic chaperone.
- Sawkar AR, Cheng W-C, Beutler E, Wong C-H, Balch WE, Kelly JW. Chemical chaperones increase the cellular activity of N370S β-glucosidase: a therapeutic strategy for Gaucher disease. Proc. Natl. Acad. Sci USA 2002; 99:15428-15433.
- Sawkar AR, Schmitz M, Zimmer K-P, Reczek D, Edmunds T, Balch WE, Kelly JW. Chemical chaperones and permissive temperatures alter the cellular localization of Gaucher disease associated glucocerebrosidase variants. ACS Chem. Biol. 2006; 1:235-251.
- Yu Z, Sawkar AR, Whalen LJ, Wong CH, Kelly JW. Isofagomine- and 2,5-anhydro-2,5-imino-D-glucitol-based glucocerebrosidase pharmacological chaperones for Gaucher disease intervention. J. Med. Chem. 2007; 50:94-100.
- Mu TW, Fowler DM, Kelly JW. Partial restoration of mutant enzyme homeostasis in three distinct lysosomal storage disease cell lines by altering calcium homeostasis. PLoS Biology 2008; 6:e26.
- Mu TW, Ong DST, Wang YJ, Balch WE, Yates JR III, Segatori L, Kelly JW. Chemical and biological approaches synergize to ameliorate protein-folding diseases. Cell 2008; 134:769-781.
- Ong DST, Mu T-W, Palmer, AE, Kelly JW. Endoplasmic reticulum Ca2+ increases enhance mutant glucocerebrosidase proteostasis. Nat. Chem. Biol. 2010; 6: 424-432.
- Ong DST, Wang Y-J, Tan Y-L, Yates JR, Mu T-W, Kelly JW. FKBP10 is an Endoplasmic Reticulum Degradation vs. Protein Folding Partitioning Factor. Chem. And Biol. 2013; 20:403-415.
- TanYL, Genereux JC, Pankow S, Aerts JMFG, Yates JR III, Kelly JW. ERdj3 is an Endoplasmic Reticulum Degradation Factor for Mutant Glucocerebrosidase Variants Linked to Gaucher’s Disease. Chem. and Biol. 2014; 21:967-976.
California: 10550 North Torrey Pines Road, La Jolla, CA 92037 - (858) 784-1000
Florida: 130 Scripps Way, Jupiter, FL 33458 - (561) 228-2000
Copyright © , The Scripps Research Institute (TSRI). All Rights Reserved.