
Transthyretin amyloid diseases
Understanding the mechanism of proteotoxicity, the tissue tropism, and why wild type transthyretin forms amyloid in ca. 10% of men, leading to cardiomyopathy.
Developing new therapeutic strategies to ameliorate neurodegeneration and organ degeneration.
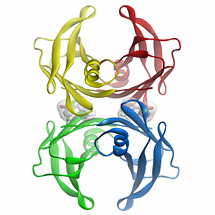
Figure 1: Three dimensional structure of the flufenamic acid-transthyretin tetramer complex. The small organic molecule, flufenamic acid, inhibits the conformational changes of transthyretin associated with amyloid fibril formation. Courtesy of Steven Johnson.
Transthyretin is a 55 kDa tetrameric protein (Figure 1) that transports L-thyroxine and holo-retinol binding protein in the blood, eyes, and cerebrospinal fluid of humans. We discovered that conformational changes alone enable transthyretin aggregation. Transthyretin amyloid formation in vivo is initiated by dissociation of the native tetramer, which is the rate-limiting step of aggregation under most conditions. Subsequently, the resulting monomers can partially denature and misassemble into amyloid fibrils and other aggregate structures, leading to the transthyretin amyloidoses, degenerative diseases wherein transthyretin aggregation leads to post-mitotic tissue loss by an unknown proteotoxicity mechanism. Aggregation of transthyretin monomers under acidic conditions (conditions that render transthyretin amyloidogenesis fast on a laboratory time scale) occurs via an efficient, or downhill, polymerization mechanism, which means that every step along the amyloid formation pathway is energetically favorable and fast relative to tetramer dissociation.
In the familial transthyretin amyloidoses, certain organs are selectively destroyed by the aggregation of mutant, and, to a lesser extent, wild type transthyretin. Why the aggregation of certain transthyretin sequences lead to the destruction of the autonomic, peripheral and central nervous systems, whereas others primarily compromise heart function is not known. The mechanism of this so-called tissue tropism is of keen interest to the Kelly lab. It is logical that these proteotoxicity differences are structure based, but there is no data to support this hypothesis. In a subset of patients, the transthyretin monomer is cleaved in half, enabling the C-terminal portion of the protein to aggregate. This could lead to different aggregate structures and tissue tropism—a hypothesis we are actively investigating.
Stabilizing the native tetrameric state of transthyretin should ameliorate transthyretin amyloid diseases: a hypothesis that is based on genetic and pharmacologic evidence. Genetic evidence is provided by interallelic trans-suppression, in which compound heterozygotes expressing both a disease-associated transthyretin mutation (e.g., V30M) and a trans-suppressor transthyretin mutation (e.g., T119M) do not develop transthyretin amyloid disease or develop very mild pathology. We have shown that incorporation of trans-suppressor (T119M) subunits into a tetramer comprised of at least one disease-associated transthyretin subunit dramatically slows dissociation of the mixed transthyretin tetramers. In other words, the tetramer is kinetically stabilized proportional to the number of T119M subunits comprising it. Pharmacologic evidence that transthyretin tetramer stabilization ameliorates the degenerative phenotypes characterizing the transthyretin amyloidoses comes from two distinct small molecule kinetic stabilizers. A potent fit-for-purpose kinetic stabilizer is tafamidis, a benzoxazole discovered by the Kelly Laboratory and developed by FoldRx Pharmaceuticals (a company that Kelly cofounded). The Kelly lab also discovered that diflunisal, a Merck NSAID available also as a generic drug in several countries, could be re-purposed as a kinetic stabilizer. In separate placebo-controlled clinical trials, tafamidis and diflunisal proved safe and effective at halting the progression of familial amyloid polyneuropathy, one of the transthyretin amyloidoses. Importantly, tafamidis provides the first pharmacological evidence that the process of transthyretin aggregation causes the transthyretin amyloid diseases—reinvigorating efforts of other investigators and companies to do the same in other amyloid diseases. A clinic trial is currently nearing completion to evaluate the efficacy of tafamidis in patients with cardiomyopathy resulting from aggregation of wild type transthyretin and/or mutant transthyretin. We are trying to understand why wild-type transthyretin aggregates in approx. 10% of men, using a variety of biophysical and cell-based experiments. Wild-type transthyretin-associated cardiomyopathy appears to be the transthyretin amyloid disease affecting the most patients.
The fit-for-purpose small molecule kinetic stabilizers of transthyretin resulted from an extensive structure-activity relationship program (in collaboration with Dr. Ian Wilson (The Scripps Research Institute, Department of Integrative Structural and Computational Biology) in which we identified and synthesized over 1000 small molecule inhibitors of transthyretin amyloid formation that grouped into half a dozen distinct families. We have shown that small molecule binding to the unoccupied L-thyroxine binding sites within transthyretin inhibits amyloid formation in vitro by selectively stabilizing the native tetrameric state over the dissociative transition state, thus raising the energetic barrier for tetramer dissociation, dramatically slowing the rate-limiting step in the aggregation pathway. The inhibitors are typically composed of two differentially-substituted aromatic rings connected by linkers of variable chemical composition. These small molecule kinetic stabilizers either just bind to transthyretin or bind and then react chemoselectively with two of the four lysines ε-amino groups within transthyretin. These SAR data allow us to predict the structures of potent and selective transthyretin amyloidogenesis inhibitors that are largely devoid of characteristics undesirable for a clinical candidate. Currently, we have a keen interest to develop second-generation kinetic stabilizers that after oral administration penetrate the eye and the brain.
In collaboration with Dr. Luke Wiseman (The Scripps Research Institute, Department of Molecular Medicine), we are investigating the relationship between the secretion of destabilized transthyretin variants and the pathology of the disease they cause. The earliest onset (onset at 20-30 years of age) and most severe transthyretin amyloid diseases are generally associated with mutations that strongly destabilize the transthyretin tetramer, resulting in facile transthyretin dissociation and misfolded monomer misassembly into aggregates post-secretion. However, the most destabilized variants characterized to date, A25T and D18G transthyretin, do not cause an early onset systemic amyloid disease because they are intercepted by the degradation component of the proteostasis network within liver cells—the liver is where most of the transthyretin in the blood plasma is produced. Because of endoplasmic reticulum-associated degradation of these highly destabilized transthyretin variants within the secretory pathway of liver cells, the concentration of the destabilized mutant transthyretin in blood plasma would not be high enough to enable the amyloidogenesis responsible for pathology. Interestingly, these mutants lead to a very rare familial transthyretin brain disease, with an intermediate age of onset (40-50 years old), because the choroid plexus is more permissive in its ability to secrete highly destabilized transthyretin variants for reasons that we are seeking to understand. These results suggest that endoplasmic reticulum-assisted folding mediated by the proteostasis network determines protein secretion in a tissue-specific manner, and we propose that its competition with endoplasmic reticulum-associated degradation may explain the appearance of tissue-selective amyloid diseases. We have recently reported arm-selective unfolded protein response activators that transcriptionally reprogram the endoplasmic reticulum proteostasis network. These activators appear to lead to selective degradation of mutant transthyretin, offering a complementary therapeutic strategy to ameliorate the transthyretin amyloid diseases.
We are also keenly interested in determining the mechanism of in trans proteotoxicity in the transthyretin amyloidoses, understanding how transthyretin fibrils are cleared from normal subjects, discerning the reason why only a subset of individuals who have predisposing mutations get transthyretin amyloid diseases, and we seek to understand the basis for tissue-selective degeneration in the transthyretin amyloid diseases. Towards these ends, we are developing methodology (e.g., peptide-based probes, conformation-dependent antibodies) to isolate non-native conformations of transthyretin that circulate in human blood and that we hypothesize cause the transthyretin amyloid diseases. We will isolate these species from patient blood and correlate their appearance/abundance with response to disease-modifying therapies. We are collaborating with the Encalada Lab (The Scripps Research Institute, Department of Molecular Medicine) which has developed C. elegans transthyretin amyloidosis models expressing a range of mutants associated with human peripheral neuropathy, cardiomyopathy or central nervous system selective amyloidosis. These models will be used to investigate the mechanism of proteotoxicity, particularly that associated with the aging process, and will enable us to test the efficacy of our transthyretin kinetic stabilizers and novel therapeutic strategies in an organismal model of the transthyretin amyloidoses having relevant neurodegenerative phenotypes.
California: 10550 North Torrey Pines Road, La Jolla, CA 92037 - (858) 784-1000
Florida: 130 Scripps Way, Jupiter, FL 33458 - (561) 228-2000
Copyright © 2025, The Scripps Research Institute (TSRI). All Rights Reserved.