|
|
|
|
|
Challenge 1: ReactivityOur lab’s research program was initiated by establishing the concept of weak coordination between common organic substrates and palladium catalysts for the palladation of C–H bonds, assisted by uniquely designed bifunctional ligands to accelerate the C–H cleavage event and thus obviating the use of exogenous directing groups. (Acc. Chem. Res. 2012, 45, 788; Acc. Chem. Res. 2020, 53, 833). This approach has led to the development of diverse carbon–carbon and carbon–heteroatom bond forming methods using C–H bonds as reaction partners for the first time.
While the combination of weak coordination and ligand acceleration has enabled a broad range of C–H activation reactions of carboxylic acids, amines and alcohols, we have also developed a complementary approach for the C–H activation of aliphatic ketones, aldehydes, (Science 2016, 351, 252) and alkyl amines (J. Am. Chem. Soc. 2016, 138, 14554; J. Am. Chem. Soc. 2018, 140, 17884) using a catalytic transient directing group strategy. The transient directing groups (TDGs) form reversible imine linkages to the substrate, thus creating a covalent intermediate highly capable of recruiting Pd for C–H activation via a L,X-type binding mode. These TDGs, rationally designed to mimic the effective ligands developed in our lab, have been widely adopted by other researchers in the field of C–H activation. Furthermore, the covalent imine linkage is well-suited for enantioinduction, as chiral information on the TDG is held in proximity to the Pd catalyst during the enantiodetermining C–H cleavage step. We have leveraged chiral TDGs to realize the enantioselective C–H arylation (Science 2016, 351, 252) and fluorination (Nature Chem. 2018, 10, 755) of aliphatic aldehyde substrates and we are working to apply this concept to aliphatic amines as well. Notably, the chiral TDG approach to enantioselective C–H activation is complementary to our chiral ligand-enabled strategy discussed below.
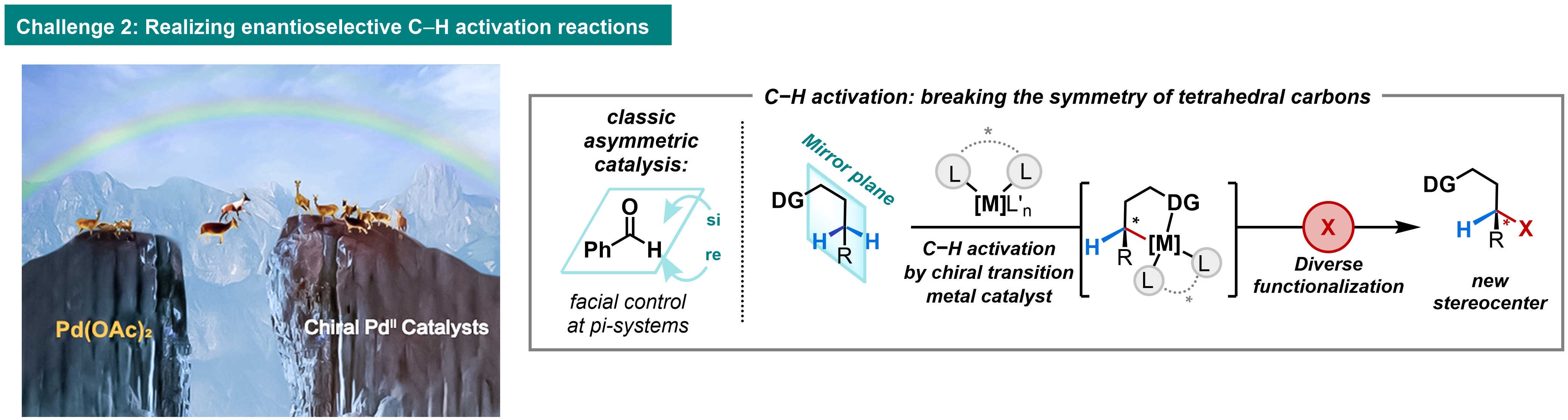
Challenge 2: EnantioselectivityOur group has leveraged the concept of ligand-accelerated catalysis to develop the first general chiral palladium catalyst for C–H activation using a monoprotected amino acid (MPAA) ligand to accelerate C–H activation and induce asymmetry under mild conditions, providing remarkable enantioselectivity (up to 99% ee) in catalytic C(sp3)–H functionalization reactions. Our transformations have been utilized in numerous total syntheses and by more than 10 pharmaceutical companies in both drug discovery and process chemistry. Building on these advances in ligand design, we have developed a diverse range of enantioselective C(sp2)–H and C(sp3)–H bond activation reactions via asymmetric C–H metalation (Science 2018, 359, 759). New generations of our chiral ligands, namely the APAQ, APAO, MPAAM and MPAThio ligand classes, are commercially available.
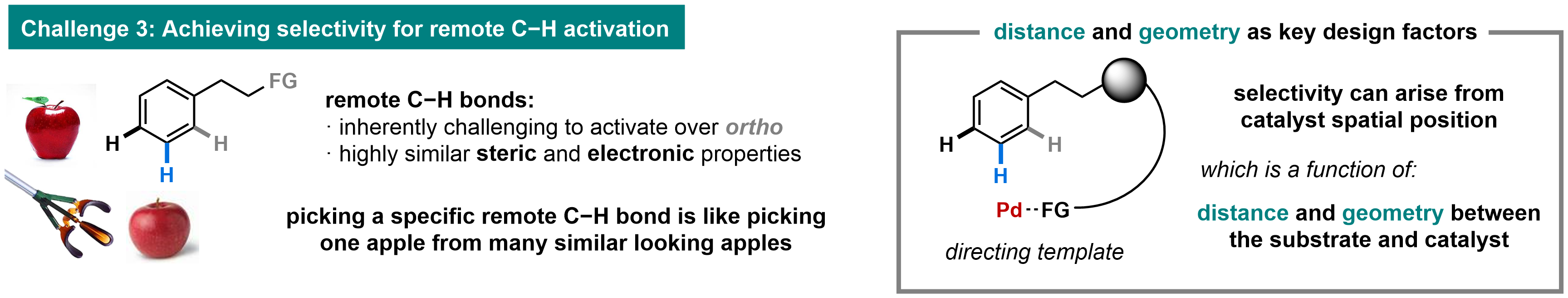
Challenge 3: Site-selectivitySite selective remote C–H activation has been another long-standing challenge in the field. Through molecular design, our group has harnessed distance and geometry as unified parameters to recognize and select specific remote C–H bonds, including those in highly similar steric and electronic environments. The initial breakthrough in achieving selective remote C–H activation reactions was based on a novel U-shaped template, favoring the activation of remote instead of proximal C–H bonds (Nature 2012, 486, 518). This finding demonstrated that distance and geometry can be employed as a powerful strategy to achieve site selectivity, overriding intrinsic electronic and steric preferences. The U-shaped templates have also been rendered catalytic via supramolecular bimetallic catalysis (Nature 2017, 543, 538). Recently, we have applied this concept of assembling macrocyclic transition states to design a Ni-based catalyst that achieves high remote site-selectivity (Nature Chem. 2021, ASAP). In addition, we have combined the proximal directing effect with a norbornene relay as a complementary approach to reach remote C–H bonds (Nature 2015, 519, 334). These discoveries have laid the foundation for realizing the ultimate objective of molecular editing: the activation of C–H bonds at any site in any order for their orchestrated functionalization. These approaches constitute a significant departure from the extensively investigated proximal ortho-C–H activation reactions and elevates the field of C–H activation to a new level (J. Am. Chem. Soc. 2020, 142, 10571).
Challenge 4: SustainabilityMost recently, a new generation of bifunctional pyridine-pyridone (PP) ligands designed to enable tautomerization between two ligand states, both critical to catalysis, has led to the first practical biomimetic C–H hydroxylation with molecular oxygen as the reagent (Science 2021, 372, 1452), as well as the dehydrogenation of aliphatic acids using molecular oxygen as the terminal oxidant (Science 2021, ASAP). These discoveries, along with other advances enabling the use of practical oxidants, including TBHP or hydrogen peroxide for C(sp3)–H lactonization, acetoxylation, and C–H/C–H coupling (Nature 2019, 577, 656, J. Am. Chem. Soc. 2021, 143, 687), have paved the way for large-sale industrial manufacturing of chemicals in a sustainable manner.
|
|
Scripps Research Institute,
10550 North Torrey Pines Road La Jolla, CA 92037 Beckman 372 |

|