
The Riewald Laboratory
Research Interest
Thrombin and activated protein C (APC) are major regulators of the blood coagulation system. Thrombin is not only the key procoagulant enzyme but it also activates the anticoagulant protein C pathway on the surface of endothelial cells. The generated APC inhibits blood coagulation by downregulating prothrombin activation in a negative feedback loop. Results from animal models and clinical trials indicate that APC has potent protective effects in systemic inflammation that are independent of its anticoagulant function and recombinant APC was recently approved to treat patients with severe sepsis. The molecular basis for APC’s anti-inflammatory effects is incompletely understood.
We have shown previously that APC signaling in endothelial cells requires binding to endothelial protein C receptor (EPCR) and activation of protease activated receptor 1 (PAR1), the thrombin receptor. PAR1 belongs to a family of seven-transmembrane G-protein-coupled receptors, which are enzymatically cleaved to expose a new extracellular N-terminus that acts as a tethered activating ligand. Thrombin cleaves PAR1 with high efficiency because thrombin directly binds to PAR1 in an orientation that favors cleavage. Thrombin-PAR1 signaling has well established pro-inflammatory effects, including disruption of endothelial barrier function, raising the question how the same receptor can also mediate protective effects of APC. Incubation of an endothelial monolayer with APC as well as very low concentrations of thrombin potently enhanced barrier integrity through PAR1-dependent transactivation of the barrier protective sphingosine 1-phosphate (S1P) signaling pathway (see Figure). The endogenous protein C activation pathway on the endothelial cell surface was mechanistically linked to powerful autocrine protective signaling by the generated APC. These results reveal an unexpected role for cross-communication between the prototypical barrier protective S1P and barrier-disruptive PAR1 pathways, and they suggest that S1P signaling may mediate protective effects of APC in sepsis. In addition, large-scale gene expression profiling demonstrated that thrombin and APC can have distinct PAR1-dependent effects in inflammatory cytokine-perturbed endothelial cells. APC-PAR1, but not thrombin-PAR1, downregulated transcript levels of several pro-apoptotic proteins including p53 and thrombospondin-1.
Taken together, our studies provide conceptually novel insight into the paradoxical condition that the two key coagulation proteases thrombin and APC, linked by a negative feedback loop, can mediate opposite effects on endothelial biology through the same receptor PAR1. The results indicate that PAR1 can mediate different biological effects dependent on rate of receptor activation and they suggest that APC elicits powerful protective responses precisely because it is a relatively poor PAR1 activator compared to thrombin. Current studies are directed at elucidating how differences in the level of receptor activation translate into activation of different cellular signaling pathways. Unpublished recent results analyzing cleavage and conformation of endogenous endothelial PAR1 on the cell surface indicate how APC-PAR1 signaling can be relevant even in the presence of thrombin. In order to dissect the contributions of PAR1 activation by APC and thrombin, PAR1 variants that are efficiently activated by APC but not by thrombin have been designed. Transgenic mice expressing these variants in endothelial cells have been generated and are currently analyzed in sepsis models to define the in vivo roles of PAR1 signaling in systemic inflammation.
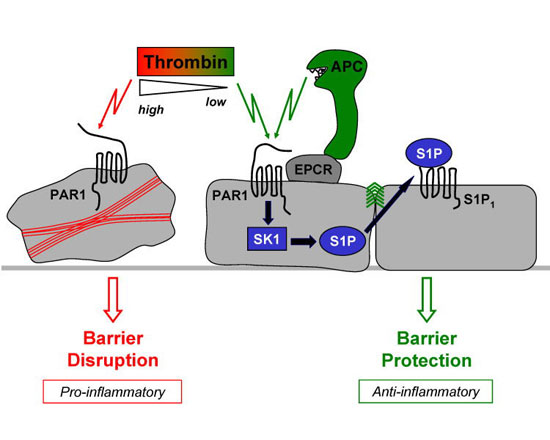
PAR1 can mediate opposite effects on endothelial barrier integrity. Inflammatory disorders such as sepsis are associated with increased permeability of the endothelial cell monolayer at the blood-tissue interface. Pro-inflammatory signaling by thrombin through PAR1 can disrupt endothelial barrier integrity. APC enhanced endothelial barrier integrity dependent on binding to EPCR and activation of PAR1, cellular sphingosine kinase-1 (SK1), and S1P receptor-1 (S1P 1). Thus, barrier protection by APC proceeds via crosstalk between the barrier-disruptive PAR1 and barrier-protective S1P pathways. Incubation of cells with very low (~40 pM) concentrations of thrombin had an equally potent S1P pathway-dependent barrier enhancing effect.
California: 10550 North Torrey Pines Road, La Jolla, CA 92037 - (858) 784-1000
Florida: 130 Scripps Way, Jupiter, FL 33458 - (561) 228-2000
Copyright © 2025, The Scripps Research Institute (TSRI). All Rights Reserved.