|
(page 2 of 2)
Ruggeri came to TSRI from Milan in 1978 for what he thought
would be a brief, one-year stay as a research associate. At
that time, he was a practicing physician who specialized in
hematology, particularly in treating patients with hereditary
bleeding disorders like von Willebrand disease.
"I came here to acquire knowledge that I could apply to
techniques to improve the diagnosis of these patients," says
Ruggeri.
He soon became interested in the mechanism of von Willebrand
disease and wound up staying an extra year before returning
to Milan. When he did return in 1980 and began working with
patients again, he found it impossible to ignore the scientific
implications of what he saw. As associate director of the
Hemophilia and Thrombosis Center at Policlinico Hospital in
Milan, Ruggeri noticed that the von Willebrand factor proteins
behaved differently in different patients with different forms
of the disease.
Von Willebrand factor, the largest protein in human plasma,
is a polymeric protein that is produced by endothelial cells
that line blood vessels. One subunit of von Willebrand factor
is more than 2,000 amino acids, and dimers of these subunits
form the building blocks of the polymer. The polymers are
large chains of various numbers of these dimers, and the size
of these polymers is related to the function of the protein,
which is to mediate the attachment of platelets to areas in
the circulation where there is a lesion. The larger the polymers,
the more sticky they become.
To induce blood clotting, the polymers should be as long
as possible, since the number of binding sites increases with
length (something biologists refer to as multivalency, with
each subunit of a multimer contributing an equal number of
binding sites).
Ruggeri observed that, in some patients, the von Willebrand
factor was not as large (and sticky) as it needed to be. In
others, it grew as large as it needed to induce blood clotting,
but it was hyper-vulnerable to attack by a blood protease
and was degraded too quickly. And sometimes, the von Willebrand
factor proteins in patients seemed to have multiple problems.
These differences in the proteins translated into differences
in how the platelets behaved—for instance, how they stuck
to one another, and how they stuck to the surfaces of blood
vessels. "The tests we were doing in [the patients] were showing
different results, and it was clear that there were different
categories of the disease," he says.
Point of No Return
Ruggeri returned to TSRI in 1982, armed with these observations
and determined to examine the causes at the molecular level.
In the years since, he has made many important discoveries
related to the disease and its various phenotypes—which,
it turns out, are caused by mutations in different parts of
the von Willebrand factor molecule.
Prior to the arrival of modern molecular biology and genomics,
this sort of work relied largely on characterizing phenotypes
and performing molecular studies that were, by today's standards,
crude. Ruggeri and others purified the von Willebrand factor
protein from plasma and digested it with enzymes into its
various domains. With the advent of molecular biological techniques
that enabled investigators to express domains of a protein
in large amounts, by the early 1990s several laboratories,
including Ruggeri's in collaboration with TSRI Associate Professor
Jerry Ware, began a line of work that has yielded several
high-resolution structures of von Willebrand factor domains.
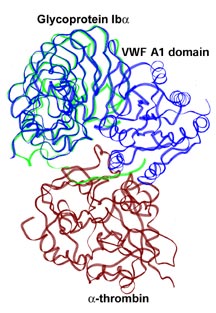
Ruggeri, in collaboration with Celikel and Varughese, solved
the first such structure, the A1 domain of von Willebrand
factor, in 1999. This was an arduous task as the A1 domain
was difficult to crystallize. As a protein, von Willebrand
factor is naturally sticky, which leads to trouble with solubility.
But once Ruggeri and his colleagues crystallized and solved
this first domain, similar studies with mutants and other
structural variants became easier.
The A1 von Willebrand factor domain has a core of beta strands
surrounded by a number of alpha helices. It appears to be
identical to domains used by a whole family of other proteins—intergrins,
for instance.
In von Willebrand factor, the A1 domain is crucial for blood
clotting because it interacts with platelets. Not surprisingly,
many of the mutations that have been reported in von Willebrand
factor protein from patients with the disease are in this
domain.
Gain of Function—In a Bad Way
Ruggeri describes how one mutation to the A1 domain von
Willebrand factor protein causes von Willebrand disease. Paradoxically,
this mutation does not knock out the function of the domain,
but causes it to become more active. The mutation causes the
A1 domain to bind to platelets too tightly.
Normally, von Willebrand factor is essential for bringing
platelets to lesions on the surface of vessels through which
blood is flowing rapidly. In order to do this, the proteins
need to bind to both the platelets and to the collagen and
other components of the matrix, which are exposed when a vessel
is cut.
"But," says Ruggeri, "they are only meant to interact [with
platelets] where there is a lesion."
The gain-of-function von Willebrand factor mutations cause
the circulating von Willebrand factor proteins to bind to
the platelets avidly, even when the platelets are in normal
circulation. The platelets, in turn, become coated with the
von Willebrand factor protein, and thus exhaust the sort of
long, sticky, multimeric polymers needed to induce clotting.
In the end, patients bleed because the platelets don't stick.
"They fly over the surface," says Ruggeri.
Ruggeri notes that several other mutations in von Willebrand
factor function in different ways to cause the bleeding disorder.
Such mutations can make the protein lose the ability to multimerize
or to bind to the platelets at all.
The Future
Knowing the molecular bases of the various causes of von
Willebrand disease is extremely helpful in designing therapies.
Patients who have the protein, but in whom it is not released
in the blood can be treated with a peptide after they are
cut. This peptide forces their cells to release stores of
the von Willebrand factor protein and corrects the bleeding
problem. This type of treatment is worthless for patients
with different kinds of mutations.
On the other hand, patients who have a malfunctioning form
of the protein per se are best treated by the administration
of normal von Willebrand factor isolated from donated blood.
Asked about the future, Ruggeri notes that there is still
a lot to do in the present. Today, he points out, even though
the A1 domain and another domain of the von Willebrand factor
protein have been solved, these account for only about one
third of the total protein. No structure for the entire protein
monomer exists and no structure for the dimer exists either.
"There is still a lot of work to do," says Ruggeri. "We
are slowly building up our knowledge."

|