
The Mackman Laboratory
Introduction
The focus of our laboratory is to study the protein, tissue factor (TF), and its effects on the physiological processes in living systems. We would like to apply the understanding of these processes to the
treatment of human diseases.
Tissue factor is the primary initiator of blood coagulation the process of blood clotting. Coagulation is a complex protease cascade involving about 30 proteins. The cascade is initiated when TF is exposed to 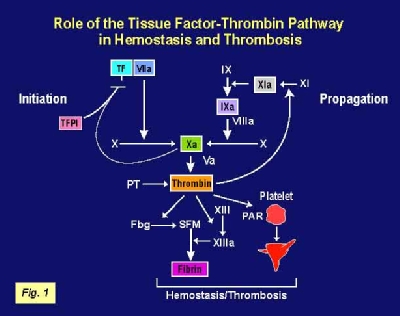 factors in the bloodstream due to vascular injury. Activation of the cascade leads to the generation of thrombin, a potent protease. Thrombin cleaves fibrinogen to fibrin which, together with platelets, form a stable clot (Figure 1). TF activity is regulated by the natural anticoagulant, tissue factor pathway inhibitor (TFPI).
factors in the bloodstream due to vascular injury. Activation of the cascade leads to the generation of thrombin, a potent protease. Thrombin cleaves fibrinogen to fibrin which, together with platelets, form a stable clot (Figure 1). TF activity is regulated by the natural anticoagulant, tissue factor pathway inhibitor (TFPI).
TF has been shown to play roles in a variety of biological processes including hemostasis, thrombosis, inflammation, metastasis and angiogenesis. The primary interest of the lab is the crosstalk between the coagulation protease cascade and inflammation in various disease models, such as sepsis, ischemia-reperfusion injury, inflammatory lung disease and atherosclerosis. We employ both genetic and pharmacological approaches to modulate levels of tissue factor, the coagulation proteases like FVIIa and FXa, thrombin and the protease activated receptors (PARs). In vitro studies have shown that thrombin activates PAR-1, PAR-3 and PAR-4. FXa activates PAR-1 and PAR-2, and FVIIa activates PAR-2. We have shown that coagulation proteases activate various PARs in different disease models, thereby enhancing inflammation.
Research Projects
Sepsis
Sepsis is induced most commonly by a systemic infection of Gram negative bacteria and the subsequent release of lipopolysaccharide (LPS) from the bacteria. The excessive stimulation of the host immune system by LPS results in high levels of inflammatory cytokines in the circulation and disseminated intravascular
coagulation. However, clinical trials of anti-inflammatory agents have not been successful in the treatment of sepsis. In contrast, several anti-coagulants have been shown to reduce both coagulation and inflammation in different animal models of sepsis. In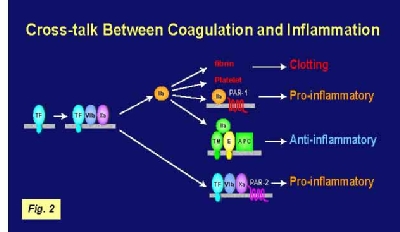 vitro studies indicated that the coagulation proteases FVIIa, FXa and thrombin activated various PARs on platelets and vascular cells. In our lab, we analyzed the role of tissue factor and PARs in coagulation and inflammation in a mouse endotoxemia model by using both genetic and pharmacological approaches. We used mice expressing low levels of TF to analyze the effects of TF deficiency either in all tissues or selectively in hematopoietic cells. Low TF mice had reduced coagulation, inflammation and mortality compared with wild-type mice. Similarly, a deficiency of TF expression by hematopoietic cells reduced LPS-induced coagulation and mortality. Inhibition of the downstream coagulation protease, thrombin, reduced fibrin deposition and prolonged survival without affecting inflammation. Deficiency of either PAR-1 or PAR-2 alone did not affect inflammation or survival. However, a combination of thrombin inhibition and PAR-2 deficiency reduced inflammation and mortality. These data indicate that hematopoietic cells are the pathologic site of TF expression during endotoxemia and that multiple PARs mediate crosstalk between coagulation and inflammation (Figure 2).
vitro studies indicated that the coagulation proteases FVIIa, FXa and thrombin activated various PARs on platelets and vascular cells. In our lab, we analyzed the role of tissue factor and PARs in coagulation and inflammation in a mouse endotoxemia model by using both genetic and pharmacological approaches. We used mice expressing low levels of TF to analyze the effects of TF deficiency either in all tissues or selectively in hematopoietic cells. Low TF mice had reduced coagulation, inflammation and mortality compared with wild-type mice. Similarly, a deficiency of TF expression by hematopoietic cells reduced LPS-induced coagulation and mortality. Inhibition of the downstream coagulation protease, thrombin, reduced fibrin deposition and prolonged survival without affecting inflammation. Deficiency of either PAR-1 or PAR-2 alone did not affect inflammation or survival. However, a combination of thrombin inhibition and PAR-2 deficiency reduced inflammation and mortality. These data indicate that hematopoietic cells are the pathologic site of TF expression during endotoxemia and that multiple PARs mediate crosstalk between coagulation and inflammation (Figure 2).
Future studies will analyze the role of TF expression by monocytes and endothelial cells in endotoxemia. Recently, we have generated HCV mice that express normal levels of human TF in an mTF -/- background. We will use these mice in bone marrow transplantation experiments to make chimeric mice: wild-type mice with HCV bone marrow and HCV mice with wild-type bone marrow. Human TF expressed by either hematopoietic (monocytes) or non-hematopoietic (endothelial) cells will be inhibited using an anti-human TF antibody that selectively inhibits human TF. Coagulation and inflammation will be measured in the endotoxemic mice to test our hypothesis that monocyte and endothelial TF play distinct roles in endotoxemia. We have also generated floxed TF mice that will permit tissue-specific knock-out of the TF gene in either monocytes or endothelial cells using mice expressing the CRE recombinase in these cells.
Ischemia/Reperfusion (I/R) Injury
Previously, we used ligation of coronary arteries in rabbits to investigate the role of TF in cardiac I/R injury. Restoration of blood flow to ischemic myocardium is associated with cardiac dysfunction and death of cardiomyocytes. This study revealed that TF expression was increased in ischemic cardiomyocytes after myocardial I/R injury. Extravascular fibrin deposition colocalized with TF-positive cardiomyocytes.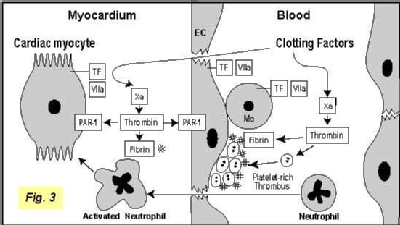 Inhibition of TF activity with a monoclonal antibody to rabbit TF administered either 15 minutes before or 30 minutes after coronary ligation reduced the infarct size by 61% and 44%, respectively. Similarly, inhibition of thrombin reduced infarct size. We have also established a model of kidney I/R injury in the lab. Currently, we are analyzing how the TF-thrombin pathway contributes to ischemia-reperfusion injury. We are investigating the relative contributions of fibrin deposition and PAR-1 signaling to I/R injury (Figure 3).
Inhibition of TF activity with a monoclonal antibody to rabbit TF administered either 15 minutes before or 30 minutes after coronary ligation reduced the infarct size by 61% and 44%, respectively. Similarly, inhibition of thrombin reduced infarct size. We have also established a model of kidney I/R injury in the lab. Currently, we are analyzing how the TF-thrombin pathway contributes to ischemia-reperfusion injury. We are investigating the relative contributions of fibrin deposition and PAR-1 signaling to I/R injury (Figure 3).
Thrombosis
In the classic model, blood coagulation is initiated when tissue factor
is exposed to FVII after vessel injury, thereby initiating clot formation (Figure 4). However, recent studies have led too the proposal of a new model in which so-called blood-borne TF contributes to the propagation of the thrombus (Figure 4). Analysis of thrombus formation in a laser-injury arteriole in real time indicated that TF present in the plasma on microparticles can accumulate on the surface of activated platelets. At present, the functional significance of this circulating pool of TF relative to that of vascular wall TF in hemostasis and thrombosis is unknown. To address this issue, we used an in vivo thrombus model. Importantly, we demonstrated that, compared to wild-type mice, our low TF mice had markedly impaired thrombus formation with significantly low TF and fibrin in the thrombus.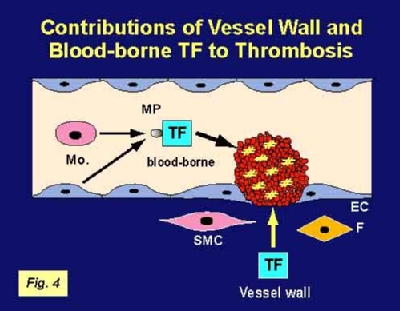 Next, we used bone marrow transplantation experiments to generate chimeric mice: wild-type mice with low TF bone marrow and low TF mice with wild-type bone marrow. Our results demonstrated that blood-borne TF is derived from hematopoietic cells and indicated that these TF-positive microparticles contributed to thrombus formation.
Next, we used bone marrow transplantation experiments to generate chimeric mice: wild-type mice with low TF bone marrow and low TF mice with wild-type bone marrow. Our results demonstrated that blood-borne TF is derived from hematopoietic cells and indicated that these TF-positive microparticles contributed to thrombus formation.
Hemostasis
Hemostasis is the physiological response to vascular injury and results in the formation of a fibrin clot that prevents blood loss from the vasculature. TF forms a hemostatic envelope around blood vessels and at body surfaces to limit bleeding in the event of injury. The TF:FVIIa biomolecular complex cleaves both FX and FIX to activate coagulation, thereby maintaining hemostasis.
One example of tissue-specific hemostasis by TF is in the heart. TF is expressed by cardiac myocytes, but not skeletal muscle cells. Recent studies in our lab have revealed the importance of TF expression in the heart. In this study, we found that mice expressing low levels of TF (approximately 1% of wild-type levels) exhibited a selective heart defect that consisted of hemosiderin deposition and fibrosis. Direct intracardiac measurement demonstrated a 30% reduction in left ventricular function in 8 month old low TF mice compared with age-matched wild-type mice. Mice expressing low levels of murine FVII (approximately 1% of wild-type levels) exhibited a similar pattern of hemosiderin deposition and fibrosis in their hearts. In contrast, FIX -/- mice, a model of hemophilia B, had normal hearts. We propose that cardiac fibrosis in low TF and low FVII mice is caused by hemorrhage from cardiac vessels due to mechanical damage and impaired hemostasis. This led to the proposal that TF expression by cardiac myocytes provides a secondary hemostatic barrier to protect the heart from hemorrhage.
California: 10550 North Torrey Pines Road, La Jolla, CA 92037 - (858) 784-1000
Florida: 130 Scripps Way, Jupiter, FL 33458 - (561) 228-2000
Copyright © 2025, The Scripps Research Institute (TSRI). All Rights Reserved.