Histone deacetylase (HDAC) inhibitors
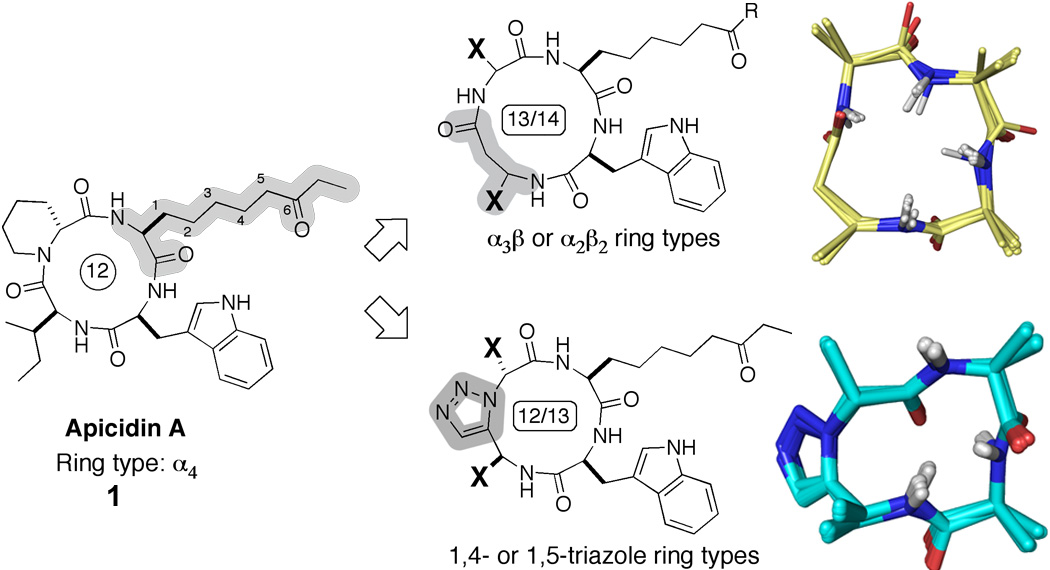
One of the principal ways cells control gene expression involves enzyme-mediated covalent posttranslational modifications histone tails in nucleosomes. Because aberrances in transcriptional regulation can lead to a variety of disease states, the development of chemical tools to specifically modulate the activity of histone-modifying enzymes could transform the ways that diseases are treated. Such chemical tools could allow us to control which genes are expressed at particular times. Our efforts in this area have focused on developing inhibitors for histone deacetylases (HDACs), which remove acetyl groups from Lysine residues in the N-terminal tails of the histone proteins. We are using rational and directed combinatorial peptide libraries to discover new inhibitors for HDACs. Using a natural product HDAC inhibitor, the non-ribosomal cyclic tetrapeptide apicidin, as lead structure, we have designed ligands that exhibit low nanomolar HDAC inhibition comparable to or better than that observed for the natural product. Our approach involved the use of backbone-modified cyclic tetrapeptides as rigid, conformationally homogeneous scaffolds for the preparation of HDAC inhibitors. These backbone modifications allowed us to simultaneously circumvent synthetic shortcomings related to the poor efficiency of cyclizing tetrapeptides, as well as to improve the conformational homogeneity of the compounds to facilitate structure determination and structure-based SAR. We have solved a series of high-resolution NMR structures for selected compounds, which has allowed delineation of three-dimensional pharmacophore elements concerning the inhibition of the HDAC activity.
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Copyright 2019 © Ghadiri Laboratory, The Scripps Research Institute |