Research
The Yates Lab focuses on Proteomics, the study of the structure and function of proteins, including how they interact with each other inside cells. This allows us to deeply investigate viruses such as HIV and SARS-CoV-2 and to measure the impact of drugs intended to ameliorate health.
The past 30 years have seen amazing advances in mass spectrometry-based proteomics technologies and their use in a broad range of biological applications. While mass spectrometry is a well-established and indispensable tool for proteomics, the Yates lab continues to strive to improve MS performance for proteomics applications and to innovate techniques that expand the scope of biological questions that can be addressed by MS.
Proteomic Informatics
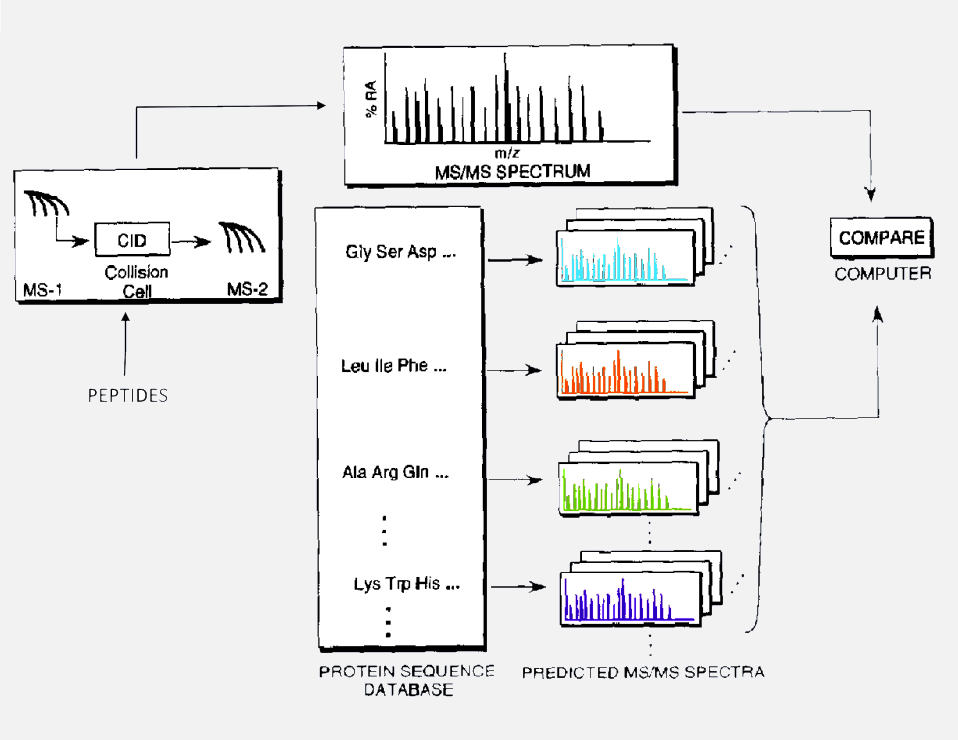
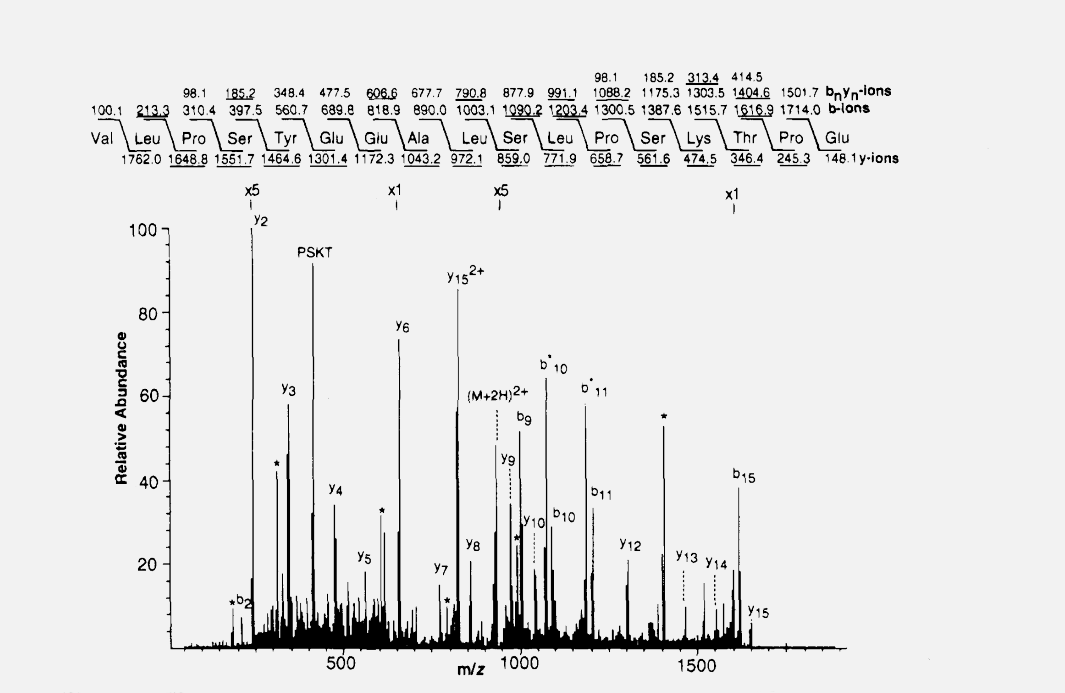
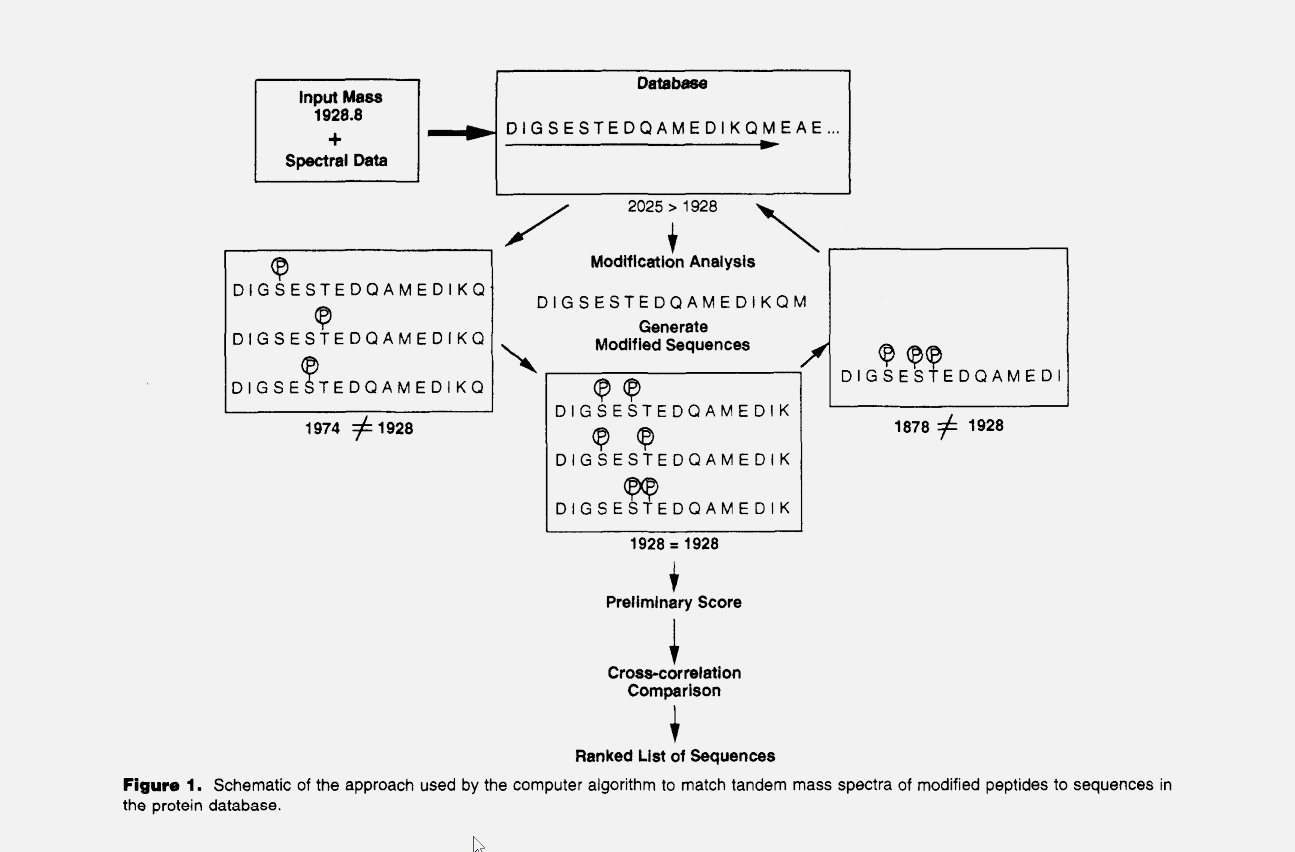
The publication of the database search algorithm SEQUEST in 1994 (Eng, J. K.; McCormack, A. L.; Yates, J. R., III. J. Am. Soc. Mass Spectrom.1994, 5, 976−989) marked the birth of the field of Proteomics. For the first time, large-scale and accurate interpretation of the tandem mass spectra of peptides was possible. This capability also created new opportunities for the analysis of proteolytically digested protein mixtures enabled by the mixture analysis capabilities of the tandem mass spectrometer. The Yates group has continued to pioneer new computational methods and tools for processing and extracting biological information from complex data sets and refining existing algorithms to keep pace with improvements in mass spectrometers and computing capabilities. These computational tools improve performance at each step of the analysis process, including the processing of raw data generated by the mass spectrometer, search methods for protein identification, protein quantification and statistical assessments of identification and quantification results.
Bioorthogonal Chemistry in Mammals
The quantification of newly synthesized proteins (NSP) at set time points using mass spectrometry has the potential to identify important early regulatory or expression changes associated with disease states. NSPs can be enriched from proteomes by employing pulsed introduction of the non-canonical amino acid, azidohomoalanine (AHA). AHA is accepted by the endogenous methionine tRNA and inserted into proteins in vivo. AHA can be enriched by reacting the azide of AHA to a biotin-alkyne (“click chemistry”). In this way, AHA containing proteins or peptides can be enriched and efficiently separated from the whole proteome. To date, the analysis of NSP has been restricted to cultured cells, but we recently developed PALM (Pulse AHA Labeling in Mammals), a strategy for quantitative tissue proteomic analysis of NSPs in animal models of disease at discrete time points through the incorporation of AHA into whole rodent proteome. Our analysis showed that less than a week of an AHA diet is sufficient to safely incorporate AHA into the proteome of multiple tissues, which allows us to identify thousands of NSP by mass spectrometry. To quantify NSPs from tissues, we devised two different labeling methods that employ the incorporation of heavy stable isotopes into biotin-alkynes and AHA molecules.
Cystic Fibrosis
Cystic Fibrosis (CF) is one of the most common and sadly, still lethal, genetic childhood diseases in the US with a median life expectancy of only 37 years. It results from mutations in the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) gene. Deletion of Phenylalanine 508 (∆F508 CFTR) is the most common mutation (affecting over 70 % of patients) and it results in misfolding and premature degradation of the CFTR protein.
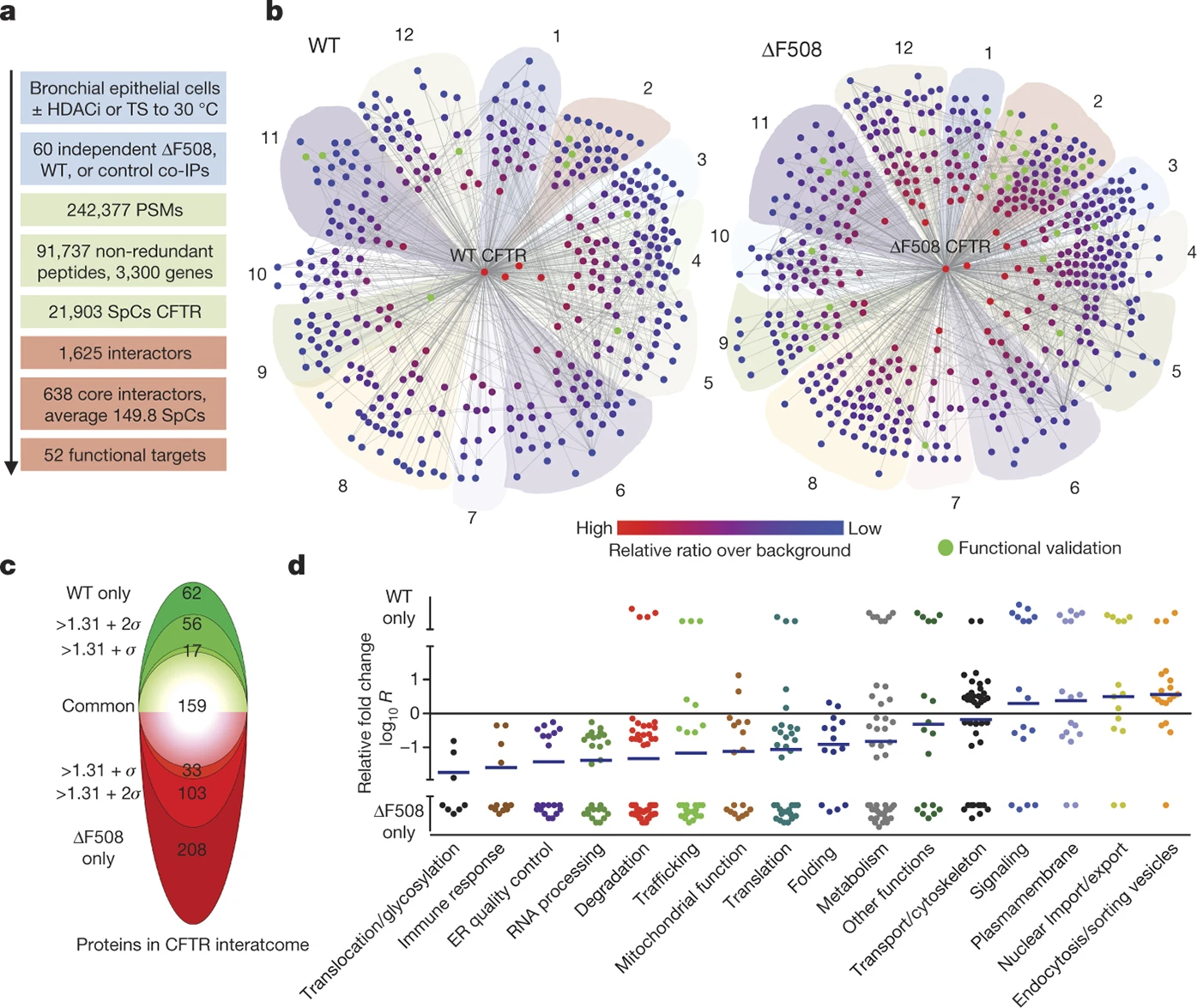
The Yates lab has made major advances in understanding the molecular defects that underly Cystic Fibrosis using mass spectrometry and ingenious method development. For example, the Yates lab deciphered the interactome of CFTR and discovered that the ∆F508 CFTR mutation leads to disease-specific protein-protein interactions that prevent proper functioning and trafficking of ∆F508 CFTR (Wang et al., Cell 2006; Pankow et al, Nature 2015; Pankow et al., Nature Protocols 2016). Their work then showed that ∆F508 CFTR function can be partially restored by blocking such disease-specific interactions in primary patient cells (Pankow et al, Nature 2015). Several of the newly discovered CFTR interactors have been further investigated by other groups as promising therapeutic targets. The Yates lab is also involved in characterizing novel lead compounds for CF therapy that were discovered in the interactome study. They also observed prominent differences in post translational modifications between wt and ∆F508 CFTR that were mirrored by other misfolded CFTR variants such as N1303K CFTR and which prevent proper trafficking of the F508 and N1303K CFTR protein (Pankow et al, Science Signaling, 2019). To help solve the puzzle of how protein misfolding affects CFTR function and trafficking, the Yates lab recently developed a novel mass spectrometry method called Covalent Protein Painting (CPP) that for the first time allowed us to characterize the structure of misfolded proteins in vivo (Bamberger, Pankow et al., JPR, 2021, Bamberger et al., 2022). By applying the CPP method, the Yates lab currently characterizes the structural defects of several CFTR mutants to help improve current and develop better future CF therapeutics.
CPP in Mammals
CPP via perfusion can preserve the in vivo conformation of proteins in their innate state, by adding dimethyl labels to exposed lysine residues on intact proteins to create a whole animal in vivo protein footprinting. Our method involves sequentially diffusing labeling reagents through blood vessels throughout the whole body so that protein surfaces are light-dimethylated [(CHD2)2] in vivo. The process is fast enough to capture protein structures in a nearly innate state. After harvest and homogenization of each tissue and lysis of the cells, proteins are denatured and proteolyzed with chymotrypsin followed by labeling of the newly exposed lysine sites with heavy-dimethyl [(13CD3)2] tags.
We applied this approach to reveal how in vivo alterations of protein conformations across tissues were associated with physiological disturbances that characterize Alzheimer’s disease (AD). We identified structural changes of co-expressed proteins and linked the communities of these proteins to their biological functions. Our findings show that structural alterations of proteins precede changes in expression, thereby showing the value of in vivo protein conformation measurement. Our method represents a new strategy for untangling mechanisms of proteostasis dysfunction caused by protein misfolding. In vivo whole-animal footprinting should have broad applicability for discovering conformational changes in systemic diseases and therapeutic interventions.
DeGlyPHER: Quantitative site-specific N-glycan analysis of vaccines against viruses
Deglycosylation-dependent Glycan/Proteomic Heterogeneity Evaluation Report (DeGlyPHER) is a sensitive, rapid, and highly reproducible method to measure broad classes of N-glycan processing and occupancy at each of the many potential N-glycosylation sites on viral spike proteins. The most promising candidates for vaccines against viruses are viral spike protein trimers, which initiate infection by interacting with host cell-surface receptors and fusing the virus particle into host cells. Heavy N-glycosylation typically shields the conserved regions of these spike protein trimers to escape host immune recognition. Viral spike proteins may comprise up to 1300 amino acids with as many as 30 potential glycosylation sites on each of their 3 protomers, so it is challenging to get the necessary near-complete sequence coverage of these heavily glycosylated proteins.
DeGlyPHER was developed in collaboration with the James C. Paulson laboratory and is being employed in two 7-year NIH/NIAID program grants awarded to a Scripps Research-led international consortium of scientific laboratories (CHAVD) to develop vaccines against HIV. DeGlyPHER evolved from methods (Cao et. al. 2017, Cao et. al. 2018) that used the "triple digestion" approach previously developed in Yates laboratory (MacCoss et. al., 2002) and sequential deglycosylation. We have recently developed it into the highly efficient "single pot" DeGlyPHER (Baboo et. al., 2021, Baboo et. al. 2023) using Proteinase K to increase sequence coverage and confidence in post-translational modifications identification (Wu et. al., 2003) and GlycoMSQuant for simpler and quicker analysis. Using this approach we have analyzed the N-glycan landscape and its dynamics on more than 200 vaccine candidates, including those against HIV, SARS-CoV-2 and its variants, Ebola, MERS, Influenza and enterotoxic Escherichia coli. The new generation vaccines against viruses are being preferentially administered as mRNA, which are expressed as spike protein trimers anchored to the viral lipid-envelope and with host-specific glycans, more closely mimicking their native state on viruses. Hence, we have adapted DeGlyPHER to analyze membrane-tethered immunogens. Many of the hundreds of vaccine candidates analyzed by DeGlyPHER have been selected for NHP (non-human primates) studies and human clinical trials, and numerous patents and publications have been submitted or are being prepared to describe these results. These pre-clinical and clinical studies have been funded by NIH and BMGF.