On Press:
Finding Selective Reversible Inhibitors In Vivo
By Jason Socrates
Bardi
Sequencing the human genome promises to be a great investment
for society. Like buying a house, the monumental public and
private effort that is the human genome promises to be profitable
over time—except the profit will be in the currency of
better medications and methods to fight diseases.
Realizing these benefits is going to take a lot of work.
We still have to do things like identify all the genes in
the genome, find out which ones are linked to particular disease
states, and find ways to modulate the effect of those genes
at the microscopic level.
Much of this work comes down to identifying small molecules
that can selectively modulate the activity of proteins that
are relevant in diseases like cancer, and certain tools are
emerging at The Scripps Research Institute (TSRI) and in laboratories
across the world that might allow us to do just that.
One of these tools is proteomics, a way of interrogating
particular cells and tissues to see which genes and proteins
might be involved in a disease. Another tool is combinatorial
libraries of small molecules, which synthetic chemists and
other scientists make (often with tens of thousands of drug-like
compounds) and then screen for potent inhibitors against the
disease-related genes identified through proteomics.
However, screening combinatorial libraries for inhibitors
generally requires purified proteins, which can be complicated
and time-consuming to produce. Also, many proteins belong
to structurally related "families," and often times inhibiting
one with a drug will inhibit another as well. There is no
guarantee that inhibitors that work well against a protein
in the test tube will not fail as drugs because they interact
with too many other, similar proteins in the body. This problem
creates bottlenecks in the discovery of new drugs.
Now, a team of researchers at The Skaggs Institute for Chemical
Biology at TSRI have combined the tools of proteomics and
combinatorial libraries in an attempt to circumvent this bottleneck.
Research Associates Donmienne Leung and Christophe Hardouin,
with Professor Dale Boger (who is Richard and Alice Cramer
Professor of Chemistry) and Associate Professor Benjamin Cravatt
have developed a proteomic method for the discovery of reversible
enzyme inhibitors from libraries of compounds.
The TSRI team has found a way to look for specific inhibitors
against particular serine hydrolases, a broad class of enzymes,
by using a technique they call competitive profiling. Competitive
profiling entails testing inhibitors against numerous enzymes
in parallel by subjecting whole proteomes to a competition
reaction between these inhibitors and activity-based chemical
probes. Inhibitors that displace probes off of particular
enzyme targets, but not other enzymes from the same family,
are identified as specific agents and chosen for further analysis
in vivo.
Using this marriage of techniques, the TSRI team reports,
in a recent article in the journal Nature Biotechnology,
the identification of reversible inhibitors of several enzymes
that bind with nanomolar affinity, including inhibitors for
the endocannabinoid-degrading enzyme fatty acid amide hydrolase
(FAAH), the enzyme triacylglycerol hydrolase (TGH), and an
uncharacterized membrane-associated hydrolase enzyme that
lacks known substrates.
This sort of approach, suggest the authors, should accelerate
the discovery of specific inhibitors against enzymes with
known and unknown function.
To read the article, "Discovering potent and selective reversible
inhibitors of enzymes in complex proteomes" by Donmienne Leung,Christophe
Hardouin, Dale L Boger, and Benjamin F Cravatt, please see:
http://www.nature.com/cgi-taf/DynaPage.taf?file=/nbt/journal/vaop/ncurrent/abs/nbt826.html

|
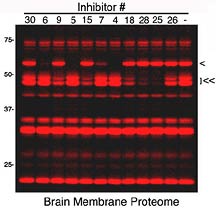
Competitive proteomic profiling of a
library of candidate serine hydrolase inhibitors with an activity-based
fluorescent probe. Inhibitor-sensitive enzymes are detected
by reduction in their fluorescence labeling intensities. Single
and double arrowheads highlight enzymes from the mouse brain
proteome that show unique inhibitor sensitivity profiles.
|

