

Fixing a misfolded protein to treat lung disease
January 10, 2023
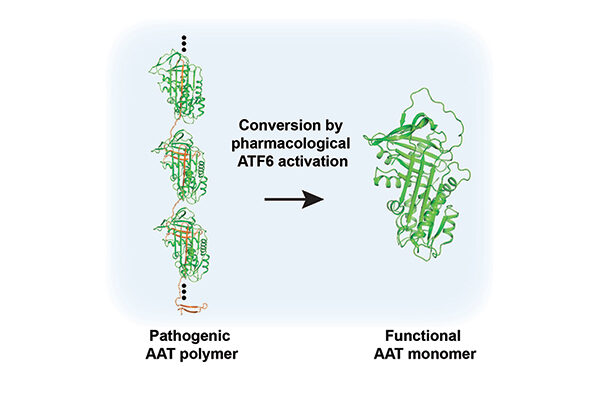
Scripps Research scientists coaxed a mutated gene into a correctly folded protein, potentially treating Alpha-1 Antitrypsin Deficiency (AATD), a genetic lung disease that affects more than 100,000 people in the United States and leads to one form of chronic obstructive pulmonary disease (COPD). Credit: Balch lab/Scripps Research
LA JOLLA, CA—Scientists at Scripps Research have discovered how to coax a mutated gene into a correctly folded protein, potentially treating Alpha-1 Antitrypsin Deficiency (AATD), a genetic lung disease that affects more than 100,000 people in the United States and leads to one form of chronic obstructive pulmonary disease (COPD). The discovery strategy based on human variation, as described in Cell Chemical Biology, works by boosting a broad-acting protein quality control mechanism already present in all cells and may be useful for treating numerous other genetic diseases.
“The road to discovery is a paradigm change,” says senior author William Balch, PhD, professor of Molecular Medicine at Scripps Research. “It’s unprecedented that a drug can be discovered that not only rescues the function of a protein, but in the case of AATD, prevents its aggregation by precisely defining the role of the quality control pathway in conversion of a misfolded state to a folded state through a broad understanding of the variation driving disease.”
AATD is a genetic disease caused by mutations in the gene encoding alpha-1-antitrypsin (AAT). In healthy people, AAT is made in the liver and travels through the bloodstream to the lungs, where it protects the lungs from inflammation and destruction. But in people with AATD, the variant AAT protein doesn’t fold into the correct three-dimensional structure when it’s first produced by liver cells. The diseased version of the protein clumps up in the liver and accumulates over many years, leading to liver damage and dangerous lung inflammation (COPD) by middle age.
When a healthy cell senses a misfolded protein, it activates the unfolded protein response (UPR). This response destroys misfolded proteins, slows the pace at which proteins are being produced, and increases levels of molecules that help proteins correctly fold. For unknown reasons, AAT variants don’t activate normal levels of the UPR.
Over the last decade, Balch’s team has developed a research method called variation spatial profiling (VSP) that aims to better understand individual disease-related proteins by analyzing how the dynamic, flexible, three-dimensional structure of proteins related to disease vary across many different people. Last year, they used VSP to discover a new therapeutic approach to address the central problem in cystic fibrosis.
In the new work, the researchers used a similar artificial-intelligence-based machine learning algorithm to study how 71 variants of AAT respond to a drug that turns up one of the three major UPR pathways in cells. Each of the 71 variants had been previously linked to AATD in human patients, and the drug had been previously developed by Jeffery Kelly and Luke Wiseman of Scripps Research.
“What we discovered is that when you manipulate the UPR, you can not only stop these AAT variants from aggregating in the liver, but you can also restore their function in the lungs,” says Chao Wang, PhD, a senior staff scientist at Scripps Research and co-first author of the new paper. “We’re able to fix the folding and activity of this protein without altering the gene sequence.”
The drug was able to correct the function of nearly all the 71 AAT variants. The researchers don’t know yet why activating the UPR works so well to coax the variants into functional conformations despite the presence of the mutation. Their current view is that when the UPR is turned-up, the environment in which proteins fold is quite different than usual. This means that even when an AAT protein contains a mutation, it’s able to be coaxed into the right functional fold, perhaps due to improved plasticity involved in shaping the molecule.
“For most AATD patients, the most severe problem is the loss of the protein in the lungs, and while many existing drugs are targeted to stop the aggregation of AAT in the liver, they do not make it functional in the lungs,” says co-first author Shuhong Sun, PhD. “So, the fact that we were able to do both is very exciting.”
In addition to pointing toward an existing compound that could be used to treat AATD, the studies also highlighted one area of the protein that is key to its function; this “gate” area of the protein needs to be able to fold and flex in particular ways for the protein to move out of the liver and do its job in the lungs. This new knowledge could lead to the development of more targeted drugs that treat AATD by manipulating this unique feature of AAT fold design. Balch’s group is already pursuing this line of work.
The success of the research also suggests that turning up the UPR may help treat other genetic diseases, including cancer and neurodegeneration in which genetic variants lead to misfolded proteins. Moreover, the conversion of pathogenic AAT fold yields new insights into the general process of natural selection in response to variation in the population.
In addition to Sun, Wang and Balch, authors of the study, “Capturing the conversion of the pathogenic alpha-1-antitrypsin fold by ATF6 enhanced proteostasis,” include Pei Zhao, Gabe Kline, R. Luke Wiseman and Jeffery Kelly of Scripps Research; and Julia Grandjean, Xin Jiang and Richard Labaudiniere of Protego Biopharma.
This work was supported by funding from the National Institutes of Health (HL141810, HL095524, AG049665, AG070209, DK123038 and AG046495) and the Alpha-1 Foundation.
Scripps Research scientists develop AI-based tracking and early-warning system for viral pandemics
Machine-learning system effectively predicts emergence of prominent variants.





Fixing the misfolded proteins that cause dementia and heart failure
All proteins have a correct way of “folding” themselves into their 3D structures. When this folding process goes awry, including processes leading to protein misassembly, a number of devastating diseases can result. In this Front Row lecture, Scripps Research professor Jeffery Kelly shared how he's developing novel therapeutic strategies to target these protein misfolding diseases, which lead to deterioration of the heart and brain. His multi-disciplinary research has already led to the development of an FDA-approved drug available in the pharmacy called tafamidis (Vyndaqel® and Vyndamax®): a medicine that slows the progression of the neurodegenerative disease familial amyloid polyneuropathy and the degenerative heart disease called TTR cardiomyopathy.

Tafamidis: A revolutionary drug for treating neurodegenerative and heart diseases
Scripps Research professor Jeffery Kelly received an unexpected visit from a patient whose life was dramatically improved by tafamidis—a revolutionary drug for treating neurodegenerative and heart diseases, developed by Kelly himself. Kelly is a leading figure at Scripps Research, spearheading efforts to address some of the most pressing medical challenges, including Alzheimer’s and various amyloid diseases. His cutting-edge research on protein misfolding has not only led to the development of tafamidis but has also broadened our understanding of protein aggregation disorders.

Front Row spotlight with Jeffery Kelly
Scripps Research professor Jeffery Kelly received an unexpected visit from a patient whose life was dramatically improved by tafamidis—a revolutionary drug for treating neurodegenerative and heart diseases, developed by Kelly himself. Kelly is a leading figure at Scripps Research, spearheading efforts to address some of the most pressing medical challenges, including Alzheimer’s and various amyloid diseases. His cutting-edge research on protein misfolding has not only led to the development of tafamidis but has also broadened our understanding of protein aggregation disorders.