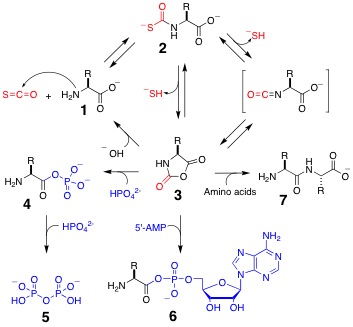
Molecular self-replication was almost certainly of central importance in the origin of life. Our lab was the first to experimentally demonstrate the feasibility of non-enzymatic peptide self-replication. We designed, synthesized, and characterized alpha-helical peptides that act autocatalytically in templating their own synthesis by accelerating the thioester-promoted amide-bond condensation of shorter fragments in neutral aqueous solution. The self-replication process displayed parabolic growth with the initial rates of product formation correlating with the square-root of initial template concentration.
We further showed that this peptide self-replication process is chiroselective. The chiroselective amplication process discriminates between structures possessing even single stereochemical mutations within otherwise homochiral sequences. Moreover, the system exhibits a dynamic stereochemical `editing' function; in contrast to the previously observed error correction, it makes use of heterochiral sequences that arise through uncatalyzed background reactions to catalyze the production of the homochiral product. These results support the idea that self-replicating polypeptides could have played a key role in the origin of homochirality on Earth.
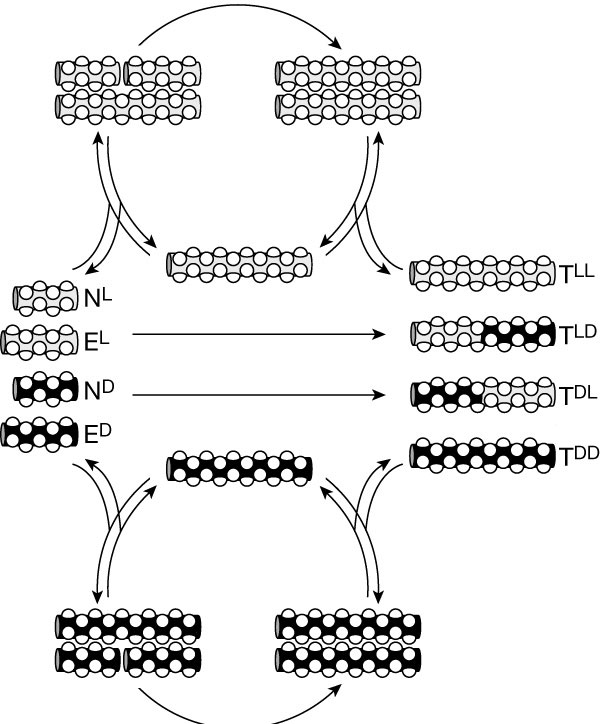
Building on the studies described above, we implemented the design, graph prediction, experimental analysis, and characterization of simple self-organized, nonlinear molecular networks. Our approach made use of the sequence-dependant auto- and cross-catalytic functional characteristics of template-directed peptide fragment condensation reactions. Starting with an array of 81 sequence similar 32-residue coiled-coil peptides, we estimated the relative stability difference between all plausible A2B-type coiled-coil ensembles and used this information to predict the auto- and cross-catalysis pathways and the resulting plausible network motif and connectivities. Similar to most complex systems, the generated graph displays clustered nodes with an overall hierarchical architecture. To test the validity of the design principles used, nine nodes composing a main segment of the graph were experimentally analyzed for their capacity in establishing the predicted network connectivity. The resulting self-organized chemical network displayed 25 directed edges in good agreement with the graph analysis estimations. Moreover, we showed that by varying the system parameters (presence or absence of certain substrates or templates), its operating network motif was altered, even to the extremes of turning pathways on or off.
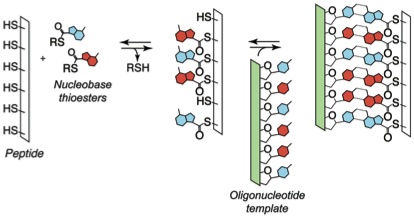
Recently, we have been exploring the central hypothesis that the dynamic nature of oligomers assembled by reversible reactions could have imbued them with advantageous features in the context of chemical evolution. We have established the viability of reversible covalent reactions in water as a basis for template-directed synthesis of sequence-adaptive peptide nucleic acids. Several classes of nucleic acid analogs have been reported, but no synthetic informational polymer had previously proven responsive to selection pressures under enzyme free conditions. We designed and characterized an oligomer family that efficiently self-assembles via reversible covalent anchoring of nucleobase recognition units onto simple oligo-dipeptide backbones (thioester peptide nucleic acids, tPNA) and undergoes dynamic sequence modification in response to changing templates in solution. The oligomers specifically self-pair with complementary tPNA strands and cross-pair with RNA and DNA in Watson-Crick fashion. Thus, tPNA combines base-pairing interactions with the side chain functionalities of typical peptides and proteins. These characteristics might prove advantageous for the design or selection of catalytic constructs or biomaterials that are capable of dynamic sequence repair and adaptation.
We are now designing, synthesizing, and characterizing novel types of peptide nucleic acids (PNA) with reversible bonds in distinct sites of their molecular structure. Our goal is to comprehensively investigate the scope and limitations of reversible reactions as a mechanism for oligomer assembly and adaptation.