A New Hypothesis about Alzheimer's Disease
By Jason Socrates Bardi
A group of scientists at The Scripps Research Institute
has proposed a new theory about the cause of Alzheimer's disease,
the progressive neurodegenerative disorder that currently
afflicts some 4.5 million Americans.
According to the hypothesis, the disease arises as a consequence
of inflammation, which creates abnormal metabolites out of
normal brain molecules.
These abnormal metabolites then modify "amyloid beta" proteins
in the brain and cause them to misfold. Misfolded amyloid
beta proteins are thought to be a major player in Alzheimer's
disease, because they can accumulate into the fibrils and
plaques that autopsies reveal in the brains of patients with
the disease. These fibrils and plaques and their precursors
are implicated in neuronal loss.
The inflammation process that creates these metabolites
can be triggered by numerous stimuli, including infections
that precede the onset of Alzheimer's disease by a significant
amount of time—perhaps years.
"If a certain inflammatory metabolite or family of metabolites
confers risk later in life, then we need to know this, and
we need to attack the problem," says Scripps Research Professor
Jeffery W. Kelly, who is the Lita Annenberg Hazen Professor
of Chemistry in The Skaggs Institute for Chemical Biology
and vice president of academic affairs at Scripps Research.
Kelly and his Scripps Research colleagues present their
new theory in an article that will be published in an upcoming
issue of the journal Proceedings of the National Academy of
Sciences.
A Progressive, Incurable Disease
Alzheimer's is a progressive neurodegenerative disease marked
by memory loss, loss of language ability, loss of the ability
to mentally manipulate visual information, poor judgment,
confusion, restlessness, and mood swings. According to the
Alzheimer's Disease Education and Referral Center, a service
of the National Institute on Aging, Alzheimer's disease is
now believed to inflict some 4.5 million people and is the
most common form of dementia among older people in the United
States. Currently, there is no cure for Alzheimer's and no
way to slow the progression of the disease.
German doctor Alois Alzheimer discovered the disease in
1906, when he examined a post-mortem patient who had died
with an unusual mental illness. Alzheimer found unusual clumps
of protein or plaques in her brain. These plaques—made
up of aggregated proteins called amyloid beta—are a clear
sign of the disease, and the aggregation of amyloid beta protein
is an accepted primary pathological marker for Alzheimer's.
But scientists have not been sure whether these fibrils
are causing the disease or are simply a marker of it. By analogy,
a tidal wave may cause massive destruction to a coastal area,
but the tidal wave itself may have been caused by a distant
earthquake undetected in that coastal area.
Kelly and his colleagues have studied the basic biology
of Alzheimer's and related diseases for many years, looking
for new treatment approaches. Now, they think they may have
taken a significant step along this path by identifying the
distant earthquake that causes Alzheimer's.
Basic Science Brings it All Together
Amyloid diseases are caused by the misfolding of proteins
into structures that lead them to cluster together, forming
microscopic fibril or plaques, which deposit in internal organs
and interfere with normal function, sometimes lethally. In
the case of Alzheimer's, these fibrils kill nerve cells in
areas of the brain that are crucial for memory.
These diseases include Alzheimer's, Parkinson's, and a peripheral
nervous system disease called familial amyloid polyneuropathy
(FAP)—a collection of more than 80 rare amyloid diseases
caused by the misfolding of the protein transthyretin (TTR),
which the liver secretes into the bloodstream to carry thyroid
hormone and vitamin A.
In the FAP diseases, mutations in the TTR protein are known
to play a direct role in causing the disease. Basically, these
mutations change the amino acid sequence of TTR, and these
changes alter protein folding in such a way as to predispose
the proteins to misfold and accumulate into microscopic fibrils,
which can then grow into the protein plaques characteristic
of FAP and other amyloid diseases.
However, in Alzheimer's disease, the cause of misfolding
is not so obvious. A number of mutations are associated with
rare forms of familial Alzheimer's, but not with most common
cases (about 95 percent of the cases). This suggests there
must be a more common cause of Alzheimer's disease, and Kelly
has combined efforts with several of his colleagues at Scripps
Research to find it.
A few years ago, Kelly started to think about traumatic
head injuries, which are a major risk factor for later developing
Alzheimer's disease. The body responds to such injuries with
inflammatory reactions that cause the release of components
of lipid membranes, such as cholesterol.
Kelly began to discuss this with his colleagues in The Skaggs
Institute for Chemical Biology, Scripps Research President
Richard A. Lerner, M.D., and Scripps Research Associate Professor
Paul Wentworth, Jr., Ph.D. Lerner and Wentworth had recently
discovered how inflammation can lead to the production of
reactive oxygen species such as ozone, which can trigger pathological
changes in other molecules in the body, like cholesterol.
In a paper last year, Lerner, Wentworth, and several colleagues
described how ozone reacts with normal metabolites to produce
toxic compounds during inflammatory processes taking place
in the body. The scientists describe two such compounds, which
they call the "atheronals." The scientists suggest these newly
identified products are critical to the pathogenesis of the
disease atherosclerosis because these atheronals were found
in atherosclerotic plaques that were surgically removed from
patients with atherosclerosis. (Atherosclerosis is a common
vascular disease that increases the risk of heart attacks
and strokes and is characterized by a hardening of the arteries
over time due to deposits of fibrous tissue, calcium, fat,
cholesterol, proteins, cells, and other materials on the inner
"endothelial" walls of an artery).
This discovery made Kelly sit up straight when he first
heard it because inflammation is increasingly seen as playing
a role in neurodegenerative diseases. Also, there are a fair
number of risk factors in common between the two diseases,
including hypercholesterolemia and inflammation.
In their new study, Kelly and his colleagues suggest that
inflammation in the brain could create a perfect storm in
which cholesterol and lipids react with ozone and other inflammatory
chemicals to produce abnormal reactive metabolites, which,
in turn, modify the folding of normal amyloid beta protein.
These modified amyloid beta proteins can catalyze misfolding
in other unmodified amyloid beta proteins, starting an avalanche
of misfolding that results—perhaps years or decades later—in
Alzheimer's disease.
A New Way of Thinking About Disease in General
To examine the hypothesis that these metabolites may be
the root cause of Alzheimer's, Kelly and his colleagues examined
the post-mortem brains of Alzheimer's patients and compared
these to age-matched controls.
They found evidence of atheronals in the brains of both
the Alzheimer's patients and the control subjects. The levels
of atheronals in the brains of the Alzheimer's patients were
not significantly elevated, but this is not necessarily surprising.
According to the new theory, the propagation of misfolding
and the buildup of fibrils inside the brain does not depend
upon continuous exposure to metabolite-modified proteins,
but to exposure during a precipitating event that may occur
a decade or more earlier. The creation of these metabolite-linked
misfolding proteins is only the initiator of the fibril plaques.
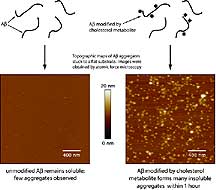
Kelly and colleagues also performed experiments in the test
tube and found that atheronals and lipid oxidation products
have the ability to dramatically accelerate the misfolding
of amyloid beta and to reduce the concentration of the protein
needed for misfolding to take place to concentrations found
in the brain.
This is an entirely new way of thinking about not only Alzheimer's
disease, but disease in general. Historically, science has
regarded disease as based on the up or down regulation of
gene expression or protein function. But this theory suggests
a new sort of pathology—the creation of a reactive metabolite
by inflammatory stress, leading to the modification of a protein,
the aggregation of that protein over time, and the degeneration
of function in the brain or whichever internal organ hosts
the aggregation.
The inflammatory metabolite theory of Alzheimer's will be
difficult to prove, admits Kelly, because the presence of
these abnormal metabolites are hard to detect years after
they initiated the aggregation. There is, so far, no smoking
gun.
"Is [this theory] right? Time will tell," says Kelly. "That's
how science works."
The article, "Metabolite-initiated protein misfolding may
trigger Alzheimer's disease" was authored by Qinghai Zhang,
Evan T. Powers, Jorge Nieva, Mary E. Huff, Maria A. Dendle,
Jan Bieschke, Charles G. Glabe, Albert Eschenmoser, Paul Wentworth,
Jr., Richard A. Lerner, and Jeffery W. Kelly and appears in
the online edition of the journal Proceedings of the National
Academy of Sciences the week of March 15-19, 2004. The
article will appear in print later this year. See: http://dx.doi.org/10.1073/pnas.0400924101
This work was supported by The Skaggs Institute for Chemical
Biology and the Lita Annenberg Hazen Foundation.
Send comments to: jasonb@scripps.edu

|