|
(page 2 of 2)
Click Chemistry
Together with TSRI Assistant Professor Valery Fokin, Professor
K. Barry Sharpless, who is the W.M. Keck Professor of Chemistry,
and Associate Professor M.G. Finn lead the second of the two
chemically oriented projects on the grant.
"Our role is to develop and synthesize molecules that could
potentially be inhibitors of HIV protease, and, using chemical
tools, to learn more about the mutations of the protease,"
says Fokin. The technique they use is in situ click
chemistry.
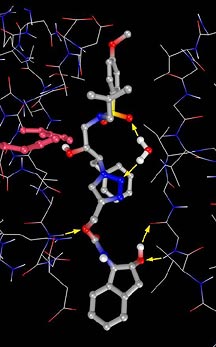
Click chemistry, a modular protocol for organic synthesis
that Sharpless developed, is a powerful and original approach
to drug design. In short, it relies on using energetic yet
stable building blocks that will react with each other in
a highly efficient and irreversible spring-loaded reaction.
In its in situ variant, click chemistry uses the target
enzyme itself to bring these building blocks together and
to direct the formation of the desired inhibitor.
The idea is to use the HIV protease itself to design its
own inhibitor by providing it with various building blocks.
Only those building blocks that can form an inhibitor will
be selected by the enzyme to "click" together. This technique
has great potential to cut through a frenzy of possible inhibitors
to demonstrate the best.
"We want to let the enzyme teach us what inhibitors [it
prefers]," says Finn. "Those, in general, should be the better
inhibitors."
The idea seems almost fantastic, but Sharpless and his colleagues
have already had success with in situ click chemistry.
A few years ago, they published a paper in the scientific
journal Angewandte Chemie describing the use of this
technique to make a powerful inhibitor to acetylcholinesterase,
a brain enzyme that breaks down acetylcholine, the neurotransmitter
that propagates nerve signals. Inhibitors of acetylcholinesterase
are used to treat the dementia associated with Alzheimer's
disease, increasing the amount of acetylcholine in the brain,
in turn enhancing brain activity. They have since expanded
on this work using this method in several other systems.
As part of the program project grant, Sharpless and Finn
want to see if they can apply the techniques of click chemistry
to designing inhibitors of HIV protease.
"That enzyme," says Finn, "should, in principle, be amenable
to the same kind of [click chemistry] strategy as acetylcholinesterase."
The strategy, Finn explains, is best applied to enzymes
that have regions of protein-protein interfaces, such as proteins
like HIV, where a dimer is formed by two identical protease
monomers. Acetylcholinesterase itself is not a dimer, but
it does have two binding regions adjacent to each other.
These protein-protein interfaces often have multiple potential
binding sites for small molecules, and the trick with in
situ click chemistry is to find classes of compounds that
will bind tightly enough in the two faces of HIV that their
proximity will allow their natural reactivity to take over.
Looking for a Short Cut
Employing the enzyme to make its own inhibitor could provide
a great short cut. For instance, to take a simple case where
inhibitors are made by combining two chemical structures—say
one of 10 "A" structures and one of 10 "B" structures—then
the possible number of structures multiplies.
With 10 possible "A" structures and 10 possible "B" structures,
there would be 100 possible compounds. But with 100 possible
"A" structures and 100 possible "B" structures, there would
be 10,000 possible compounds.
"We don't have to make and screen all those," says Fokin.
"We just have to allow the enzyme to select which ones fit
best."
Inside the enzyme's binding pocket, the components should
click together into a potent inhibitor of HIV protease, and
once Sharpless, Fokin, and Finn recover the inhibitor, they
can determine its structure and produce it in much larger
quantities so that their colleagues on the program project
grant can study the interaction of this inhibitor with the
protease in structural and tissue culture experiments.
As the cycle continues, the way that the TSRI researchers
envision it, other members of the project will provide mutant
and wild type protease, and the Sharpless, Fokin and Finn
laboratories will then repeat the process.
Dynamic Therapy
At the moment, this project is still in the early stages.
Together with the computational team, Sharpless, Finn, and
Fokin are deciding on the best building blocks to start with.
The Sharpless group has also been developing a benign copper
catalyst and a methodology for copper-catalyzed "stitching"
of azide and alkyne building blocks that will allow them to
make a variety of the inhibitor analogs they are interested
in. In addition to generating libraries of the analogs, the
technique could also be used to produce large amounts of click
inhibitors for further studies.
"Everything changed when we discovered the copper-catalyzed
process for the synthesis of triazoles," says Fokin. "It makes
the whole process go a lot smoother."
Another intriguing question they are asking is whether they
can possibly apply the technique of in situ click chemistry
in vivo.
In vivo means literally "in life," which in biology is generally
understood to apply to experiments that take place in a living
organism. In this case, in vivo in situ click chemistry
suggests the development of a new type of cocktail—one
that is made inside the target enzymes inside a living organism.
The idea is to give a cocktail of building blocks to a patient
from which the final structure of the best inhibitor will
be made. Only those building blocks that are effective against
the protease enzymes encoded by the particular strain of HIV
that infects that one patient would react and make inhibitors.
Another intriguing alternative is to provide a pharmacist
with a collection of building blocks, or "pre-drugs", which
can be dispensed to each patient based on the specific information
about the mutation.
"We asked, 'Can we actually apply click chemistry to treatment?'"
says Fokin. "'Can we use our bodies to decide what kind of
inhibitors to make?'"
The idea is that the therapy would be flexible enough so
that it would work no matter which strains of HIV infect a
person. By giving patients the subunits, the particular drug
needed at that moment would be selected by whichever strain
of HIV infected them.
While these ideas are tantalizing, they are a long way from
becoming a reality. The investigators are still doing the
first in situ studies with HIV, and any eventual in vivo
studies would have to be done in cells first and then in model
systems, before extensive human trials for safety and efficacy
could even begin.
Still, says Fokin, the concept as they envisioned it offers
a new way to approach what is now the long-standing problem
of HIV drug resistance.
1 | 2 |

|