When Paths Collide—Redundancy, Angiogenesis, Cancer,
and Combinatorial Therapy
By Jason Socrates
Bardi
Redundancy may be a classic mark of poor writing, but in
other crafts, redundancy is not always, always a bad
thing.
Engineers use redundancy as a paradigm to ensure safety
in design, and redundancy has evolved in biological systems
for apparently the same reason. Having more than one copy
of the same gene, for instance, means that people who inherit
a mutant form of a certain gene from one parent might still
live healthy lives with a healthy gene from their other parent.
But when a set of normal biological processes go awry, redundancy
can confound the best efforts of science.
Such is the case in cancer. For years scientists have sought
to fight cancer by designing compounds that block angiogenesis—the
proliferation of blood vessels that often accompanies the
growth of solid cancer tumors. These new blood vessels bring
necessary nutrients and oxygen to the hungry tumor cells.
Block angiogenesis, the thinking goes, and you can starve
a tumor—like drying out a lake by diverting all its tributaries.
But the body has more than one way of proliferating blood
vessels. A number of years ago, several scientists including
Professor David Cheresh, a member of the Department of Immunology
at The Scripps Research Institute (TSRI), described how angiogenesis
can be initiated by different growth factors, including vascular
endothelial growth factor (VEGF) and basic fibroblast growth
factor (bFGF).
Significantly, VEGF and bFGF can both induce angiogenesis
in tumor cells by coordinating their activities with different
"integrin" cell adhesion receptors. VEGF acts with the integrin
protein alpha(v)beta(5), and bFGF acts with the integrin alpha(v)beta(3).
Designing drugs to target only one of these growth factors/integrin
pairs may not be completely effective at stopping angiogenesis
or tumor growth because a cancerous cell might simply use
the other pathway.
But the two pathways are not entirely distinct from one
another.
In a paper published in a recent issue of the journal Science,
Cheresh and a team of scientists at TSRI have described how
these two pathways intersect in the activation of a protein
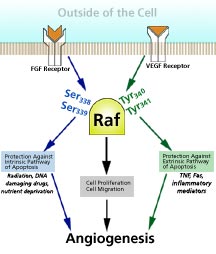
called Raf—a "kinase" enzyme that is involved in signal
transduction. Cheresh and his colleagues describe how endothelial
cells use VEGF and bFGF as survival factors to activate Raf
as a way of protecting themselves against distinct inducers
of apoptosis (programmed cell death) in order to survive—during
tissue remodeling events associated with wound repair, inflammation
or cancer.
Interestingly, activated Raf does two different things in
the cell depending on whether it is activated by VEGF or bFGF.
When Raf is activated by bFGF, the activated Raf is driven
to the cell's mitochondria, where it protects the cell from
stress-mediated death, providing protection against radiation
or chemotherapy, which would normally cause a cell to undergo
apoptosis.
When Raf is activated by VEGF, on the other hand, the activated
Raf protects the cell against receptor-mediated apoptosis,
which happens at a site of inflammation, for instance, where
the body's cells release chemicals like tumor necrosis factor
(TNF) or Fas, which can bind to receptors on otherwise healthy
cells and induce cell death. These two pathways of endothelial
cell survival probably exist to ensure angiogenesis can take
place among remodeling tissues within distinct microenvironments,
says Cheresh.
Importantly, tumors induce a blood supply by activating
both of these angiogenic pathways. Consequently, Cheresh and
his colleagues' findings have a significant impact on how
to attack the tumor blood supply: blocking Raf activity might
promote endothelial cell death regardless of the angiogenic
stimulus.
This idea further validates Raf as a fundamental target
for the design of drugs for treating cancer. A year ago, Cheresh
and his colleagues published another paper in Science
describing an experiment in which they delivered a mutant
form of the Raf gene to tumor-bearing blood vessels. The mutant
Raf caused endothelial cells within the tumor to die, which
was then followed by tumor cell death and regression of large
preexisting tumors.
Rather than targeting the growth factors or their receptors,
a drug might be designed to target Raf—as a single "bottleneck"
downstream of VEGF and bFGF, thereby ensuring blood vessel
death regardless of the angiogenic stimulus.
In addition, cancer cells probably use Raf for their own
survival in a similar way. Tumor-cell-associated Raf can be
activated either by growth factors within the tumor microenvironment
or oncogenes within the tumor cells themselves. Once activated,
Raf can promote one or both of the pathways leading to cell
survival. Therefore, besides validating Raf as a target to
block angiogenesis, this work also points the way for designing
strategies for combining existing anti-cancer drugs to attack
tumors directly.
Combinatorial drug therapy is a treatment paradigm that
has proven effective in diseases ranging from AIDS to cancer.
But the key, of course, is knowing which drugs to combine.
In this case, understanding that Raf in tumor cells can promote
survival in the face of distinct inducers of cell death will
make it possible to either target Raf directly or combine
drugs to effectively ensure that both apoptotic pathways are
active. For instance, one might combine a drug like Taxol
that induces stress-mediated death with one that promotes
receptor-mediated death.
"The combination of those approaches might prove to be very
potent or synergistic," says Cheresh. "We think these studies
will impact our ability to design and use drugs to block angiogenesis
and will also help design and use combinatorial drugs against
cancer directly. However, for now we are focusing most of
our efforts on targeting Raf itself, which appears to be a
key enzyme that proliferating endothelial cells and tumor
cells use to stay alive."
To read the article, "Role of Raf in Vascular Protection
from Distinct Apoptotic Stimuli" by Alireza Alavi, John D.
Hood, Ricardo Frausto, Dwayne G. Stupack, and David A. Cheresh,
please see the July 4, 2003 issue of the journal Science,
or go to: http://www.sciencemag.org/cgi/content/abstract/301/5629/94.

|