|
(page 2 of 2)
The Structure of Viruses
Brooks and Case also collaborate on a National Institutes
of Health (NIH) center to develop multi-scale modeling tools
for structural biology and to make these tools available to
the scientific community for free.
"Our mandate is to build tools to help structural biologists
better interrogate, explore, and understand structural models,"
says Brooks, who is director of the NIH center.
One of the center's objectives is to develop tools that
might be used for genome-scale modeling and structure prediction.
As more and more genomes are solved and annotated with gene
prediction algorithms and proteomic techniques, the number
of proteins of unknown structure and function is growing,
which is creating a great demand for computational tools that
can predict the structures—or partial structures—of
these proteins.
Brooks and Case work on tools for problems including protein
folding prediction and homology modeling—where the structure
of a protein is predicted based on the similarity of its amino
acid sequence to another, known protein—and test these
tools for their ability to predict the fold of unknown proteins.
The computations that they use are sometimes very intensive.
"We recently ran a calculation in which we used 2,500 nodes
(processors) at the Pittsburgh Computing Center together with
about 500 nodes of San Diego's machine and hundreds of nodes
as a site in Virginia," says Brooks. "They were all working
at once for one computation for a period of 24 to 48 hours."
That particular calculation, says Brooks, involved exploring
a protein-folding landscape for a protein that is known to
fold very quickly in order to understand how the sequence
of amino acids in the chain determines the three dimensional
structure of the folding protein.
In another area of research at the NIH center, Brooks and
Case aim to make connections between atomic-level descriptions
of molecules obtained from crystallography and NMR and lower-resolution
pictures obtained with other techniques, such as electron
microscopy (EM).
This is important because in solving structures, crystallographers
and NMR spectroscopists often can only solve a small piece
of a large structure, while EM can handle large structures,
but not at high-resolution. Reconciling the two allows them
to fit high-resolution pieces together, jigsaw-like, in order
to obtain a complete picture with more atomic detail than
would be possible using only one technique or the other.
They then take this still-static picture and make a sophisticated
model out of it, modeling the dynamics of the molecule at
a larger scale.
"Generally, the reason for doing this," says Case, "is to
get biologically interesting states that are not observable
at high resolution.
They have also worked in collaboration with TSRI Professor
Jack Johnson to study the assembly of viruses—a subject
that Johnson has been studying with x ray crystallography
for a number of years.
"We're looking at understanding, at a molecular level, the
process that swells and shrinks the particles," says Brooks.
Virus particles are dynamic in solution and undergo large
structural changes throughout their "lifetime," and some of
these changes are interesting biologically because they may
be related to such issues as how the virus particle gets its
genetic information into a cell that it infects. However not
all the changes may be accessible experimentally, since the
transition states may be unstable and therefore impossible
to crystallize or study with NMR.
Brooks and Case are also trying to understand how nucleic
acids are packaged in viruses. "Usually that cannot be seen
at high resolution," says Case, "because the DNA or RNA is
too disordered."
Still, he adds, the problem is not so simple computationally
either. The insides of a virus are incredibly crowded, which
makes computing difficult. Some of the work they do involves
figuring out how to remove atomic detail from the structures
so that they do not have to take it into account. Case, for
instance is working on how to model a protein or DNA in continuum
solvent to simplify the calculations.
"You keep an atomic-level protein or DNA model, but you
remove all the atomic level descriptions of the water or ions,"
he explains.
The Fluctuating Ribosome
Brooks is particularly interested in the workings of the
large molecular machines that carry out much of the work of
the cell, such as the ribosome or actin/myosin.
"Nature effectively exploits the shape of these objects
to provide robustness in the motions that they have to undergo,"
says Brooks.
In the case of the ribosome, the starting point for the
study of these motions are the near atomic-resolution and
atomic-resolution molecular structures that have been solved
in the last couple of years using EM and x ray crystallography.
Brooks is working to create atomic-level models using these
structures that give dynamic movement to them. He, with his
postdoctoral collaborator Florence Tama, is building an elastomechanical
model that captures the shape of molecular "objects" and allows
dynamic behavior to emerge from normal vibrations and rotations
associated with the atoms in the molecule.
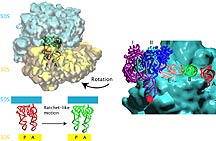
These dynamic movements may be the key to some of the molecules'
most complex behavior. In the model of the ribosome, the collective
fluctuations may give rise to a ratchet-like motion that is
involved with the phenomenon known as translocation.
Translocation is an important part of the ribosome function
because it involves moving tRNA molecules loaded with amino
acids from the site on the ribosome where the anticodon of
the tRNA is paired with the codon of the mRNA to the site
on the ribosome that catalyzes the formation of the peptide
bond between the amino acid loaded on the tRNA and the growing
protein chain. A major motion of the ribosome is associated
with this movement from one location on the ribosome to another
after an "effector" molecule binds to the ribosome.
In Brooks' dynamic model of the ribosome, the movement occurs
quite naturally, as one of the "normal modes" of vibration.
Physicists describe normal modes as fluctuations about a local
energy minimum (preferred vibration) for simple harmonic oscillations—between
two adjacent atoms, for instance. More collective motions
in large molecular assemblies like the ribosome may describe
functionally important dynamics.
"[The translocation] is happening only because of the shape
of the molecule," says Brooks. "It seems as though nature
somehow engineers these shapes so that it is a single mode
that is functionally relevant."
These models are promising because they may be the only
way to access atomic-level structural information about transition
states of large structures like the ribosome. Such transition
states may be inherently unstable and completely inaccessible
to experimental techniques but nevertheless important to the
operational cycle of these cellular machines—in other
words, they may hold some of the secrets of life.
And perhaps of physics as well.
1 | 2 |

|