

Trapped in the Folding Machine
By Eric Sauter
For the last decade or so, William Balch, a professor in the Department of Cell Biology and the Institute for Childhood and Neglected Diseases at The Scripps Research Institute, has been working with the endoplasmic reticulum (ER), a seemingly endless network of tubes and sacks within the cell cytoplasm, and a special place for proteins destined for different locations inside and outside the cell.
"The fact is that the ER is the origin of many proteins," Balch said. "You have in your body about 35,000 proteins encoded by the genome and about one third of those are transmembrane proteins or lumenal proteins that proceed to their final destination via the ER. At any given time, in the average cell there may be thousands of different proteins competing for transport from the ER."
In the ER, the actions of chaperones, highly abundant and sometimes finicky protein helpers, aid in a major way in guiding the folding of proteins during the first step in their production, synthesis by ER-attached protein synthesis factories called ribosomes.
In a major new study "Hsp90 Co-Chaperone Aha1 Downregulation Rescues Misfolding of CFTR in Cystic Fibrosis," recently published in the journal Cell, Balch and colleagues focused on one protein and one chaperone helper in particular. The study represents a milestone in the field and may provide a framework for the correction of the childhood disease cystic fibrosis and other protein folding diseases.
An Inherited Disease
The primary, crippling symptom of cystic fibrosis, an inherited disease that begins in early childhood, is abnormally thick mucus that blocks airways and causes difficulty breathing. Associated symptoms include incapacitating lung infections, intestinal disorders, diabetes, and infertility. About 30,000 people in the United States have cystic fibrosis, according to the Cystic Fibrosis Foundation. An additional 10 million more—or about one in every 31 Americans—are carriers of the defective gene, but do not have the condition.
Cystic fibrosis is triggered by a genetic mutation that causes the defective folding and retention of a specific protein—cystic fibrosis transmembrane conductance regulator (CFTR)—in the endoplasmic reticulum.
"In this case, we're not studying a nice compact soluble protein that gets secreted," Balch said. "CFTR is a very complicated multi-membrane spanning protein crossing the lipid bilayer many times, with the different linked parts (called domains) found on different sides of the cell membrane. These are brought together by sophisticated pathways mediated by chaperone 'folding helpers.'"
The CFTR protein acts as a membrane channel for control of movement of chloride ions between the interior and the surface of the cells lining the ductwork of the body. The channel also interacts with and regulates the function of sodium channels, thereby playing a key role in controlling the balance of salt between the inside and outside of the cell. Maintaining the right salt balance across the cell surface is critical for water balance throughout the body, especially in places like the lungs, pancreas, and gastrointestinal tract where a high level of surface flow is necessary for normal function.
When the body's system is working correctly, the CFTR protein is properly assembled in the ER. Here, ribosomes synthesizing the protein cooperate with the helper chaperones. Chaperones come in different flavors; certain chaperones are specialists and like to work with certain types of proteins, while others don't care whom they work with (the generalists). Generalist chaperones bind the protein in the unfolded state, stabilize it, and, once the protein has gone through the complete folding event (often assisted by the specialists), release it for transport to the cell surface.
In the case of CFTR, both the generalist heat shock protein Hsp70 and specialist Hsp90 families work together to assemble the final product. Hsp70 and Hsp90 are extremely abundant, each comprising about two to three percent of cytosol, which is the fluid part of the cell surrounding the ER. The CFTR protein leaves the ER and works just fine on the cell surface if it's folded correctly, a process that in nature takes approximately ten minutes. If not folded correctly, the CFTR protein doesn't get to leave the ER.
"What's Wrong Here?"
In cystic fibrosis, that's where everything starts to go wrong. A single, common mutation in the CFTR gene, found in 70 percent of patients, results in the loss of a single amino acid in just one domain. As a result, when the chaperone helpers try to assemble the protein, Balch said, they do not see the protein as they should. Instead, the mutant CFTR is better recognized by a degradation system designed to detect misfolded proteins in the ER. This degradation pathway sends CFTR into the waiting arms (or jaws) of the proteasome.
"If it misfolds, the protein gets dragged into the proteasome, a cylinder filled with proteases, where it gets chewed up," Balch said. "The proteasome is like a garbage can with teeth."
During early childhood when CFTR fails to populate the surface of cells in the lung, pancreas, and intestine, the results are horrendous. The salt balance is upset, the mucus on the cell surface becomes thick and sticky, and that sets off the cascade of events that is cystic fibrosis. The salt imbalance kills lubrication in the lungs, which encourages infections and triggers inflammatory responses, often leading to pneumonia which becomes harder and harder to treat.
But why should the loss of a single amino acid cause such a terrible response?
"That's the big question, 'What's wrong here?'" Balch said. "My first response is we don't know the full answer. But we do know that the body somehow decides that the protein is destabilized, the protein is energetically not able to get through a step in the folding pathway guided by helper chaperones, and the degradation pathway takes over."
Intriguingly, unlike the wild-type protein, mutant CFTR appears trapped in the folding machine.
In the new study, the researchers took a closer look at the entire chaperone helper system.
For this study, the Balch laboratory collaborated with the laboratory of Professor John Yates, a fellow faculty member of the Department of Cell Biology. Yates applied his revolutionary, state-of-the-art shot-gun mass spectrometry approaches (referred to as multi-dimensional protein identification technology or MudPIT) that looks at thousands of proteins at a time.
"The results surprised us," said Balch. "They revealed a remarkable diversity of molecular chaperones and co-chaperones required to control the folding and stability of CFTR during synthesis."
In fact, their studies suggested that a collection of both Hsp70 and Hsp90 regulators, involving potentially up to 40 different proteins, normally help prepare and fold CFTR for export to the cell surface. After the generalist Hsp70 family chaperones have completed early folding steps (as previously suggested by other researchers studying CFTR), it appears that the specialist Hsp90 system completes the project. Hsp90 likes to work with mostly-folded proteins—indeed, up to 600 "clients" have been identified in the cell, for which Hsp90 either completes the fold or allows them to bounce back and forth between folded and partially folded states, turning on and off function.
"The Hsp90 chaperone is like the relief pitcher that comes on in the ninth inning to finish the game after the other pitchers have slogged through the rest of it," Balch said. "The Hsp90 chaperone is a finisher, the game closer, and thus critical for function."
Others have noticed this ability as well. Several companies are eyeing the commercial potential of Hsp90 in control of cancer cell growth because a number of Hsp90 clients control cell propagation.
Balch and others are hoping to capitalize on Hsp90's unique activity in folding to correct cystic fibrosis. In the recent study, Balch and Yates report that the activity of one component of Hsp90 system can be adjusted to promote delivery of the mutant protein to the cell surface, where it has chloride channel activity. This also leads to a new view of the energetics of the folding environment in the cell, an area being investigated in collaboration with the laboratory of Professor Jeffery Kelly, an investigator in the Scripps Research Department of Chemistry and the Skaggs Institute of Chemical Biology.
"Because the Hsp90 system often adjusts the final steps of folding, we will be looking to identify small molecule compounds that now assist this pathway and give the mutant CFTR protein a second chance to escape the misfolding step," Balch said. "We're on a path that may eventually lead to an understanding of how a misfolded protein can be turned into a more folded protein with small molecule drugs."
A Pivot Point
Cystic fibrosis isn't the only misfolding disease that could be illuminated by this work—other protein-folding diseases include Alzheimer's disease,Parkinson's disease, Creutzfeldt–Jakob disease, Huntington's disease, sickle cell anemia, Tay-Sachs, and Gaucher's disease—but cystic fibrosis is the one that has caught Balch's attention.
He is aware that other approaches to correcting the error haven't worked very well. Gene therapy, once held out as the answer to cystic fibrosis, has been disappointing to date. Moreover, the many common therapeutic approaches pioneered by the Cystic Fibrosis Foundation, while important advances for the patient in terms of life-style and longevity, do not address the root cause of disease and require a life-long dedication to a regimen of pharmacological and physical interventions.
"You can try to hit the downstream problems with, for instance, antibiotics and anti-inflammatory therapeutics, but eventually you run into a number of intractable conditions given the progressive onset of disease," Balch said. "Our approach and the current thinking by others in the field is to adjust the fold or the folding environment so that the body sees the mutant CFTR more as a 'polymorphism'—an acceptable difference from normal activity—and therefore exports it to the cell surface where it can function. While others in the field are now beginning to successfully identify drugs using 'high-throughput screens' (cell-based assays that test hundreds of thousands to millions of compounds for their ability to promote delivery of the chloride channel to the surface), our study now provides important hints as to the mechanisms involved that might allow this to happen."
What's important, Balch said, is that by adjusting the local chaperone helper environment, you could get exceptional results.
"For children with cystic fibrosis, the current thinking is that if you can get five to ten percent of the wild type or naturally occurring level of the CFTR protein to the cell surface, the kids would be much better off. This is because there are over 1,000 mutations in this disease with different levels of defects—some of these kids have a mild form of the disease, yet only a very low amount of CFTR at the surface. It may be that for the most common form of the disease, all we have to do is inch the folding system over a bit, making it more generous or accepting of these mutations."
A Broad-Based Front
With support from the National Institutes of Health and the Cystic Fibrosis Foundation, coupled with his long-standing collaborations with Scripps Research colleagues Yates and Kelly, Balch and his colleagues are working to create a broad-based front to ask and answer the most challenging questions about protein folding and misfolding.
Balch is a co-director of a consortium recently initiated by the Cystic Fibrosis Foundation to bring investigators together in a forum dedicated to a wide range of scientific interactions focused on the folding problem. As part of that effort, the researchers are working to shrink the entry barriers to this research by generating reagents that will be freely available to the entire academic community, a project also funded by the Cystic Fibrosis Foundation. In addition, the group plans on a central web site, which will hold a comprehensive database on pathways that encompass CFTR function.
Balch hopes the information network will be able to bring together the many separate and important observations generated by investigators in the cystic fibrosis field into a broader, systems-wide view that will lead to advances that make a difference in patients' lives.
"We think that the 'CFTR interactome' described in the Cell paper is an important starting point to understand the origins of cystic fibrosis disease," he said. "We've now generated a preliminary roadmap for us and others to further develop. As a community, we are now trying to make the biology work for us in correction of disease. If we can solve the CFTR folding problem, the first step in the pathway, the many downstream irregularities that trigger disease pathology could click into place, solving many problems that are currently treated on a one-by-one, daily basis."
Send comments to: mikaono[at]scripps.edu

"We think that the 'CFTR interactome' described in [our recent] Cell paper is an important starting point to understand the origins of cystic fibrosis disease," says Professor William Balch.
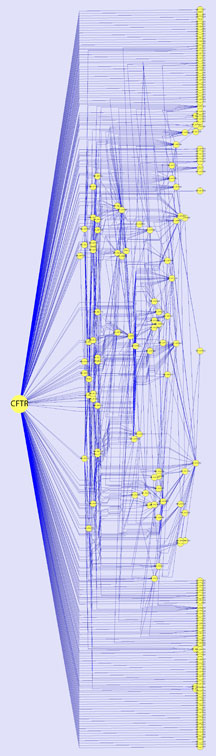
The "CFTR interactome" comprises a network of many proteins contributing to the normal folding, trafficking, and function of the protein CFTR, which has been implicated in the disease cystic fibrosis. Click for details
