

Wet Lungs or Dry?
Scientists Discover New Key to Pulmonary Edema in Respiratory Distress Syndrome
By Jason Socrates Bardi
Physiology is sometimes a crossroads where many different paths converge. Such is the case with acute respiratory distress syndrome, a severe and often fatal condition also known as adult respiratory distress syndrome or simply "shock lung."
Acute respiratory distress syndrome can be caused by a number of underlying conditions, including smoke inhalation, a severe blow to the chest, bad pneumonia, septic shock, severe blood loss, or drug overdose. Although the causes vary greatly, the situation for a patient who arrives at an emergency room with acute respiratory distress syndrome is largely the same—critical. Adult respiratory distress syndrome leads to the filling of the lung's airways with fluids, a condition known as pulmonary edema. This leads to a reduction of oxygen intake, which can rapidly degenerate into complete respiratory failure.
"It's a serious complication that often results in death," says Professor Hugh Rosen of The Scripps Research Institute.
Adult respiratory distress syndrome is usually treated by ventilation that increases the oxygen available to the lungs, as well as by antibiotics, muscle relaxers, pain relievers, heart stimulants, and other drugs that address some of the related problems. According to the U.S. National Heart, Blood, and Lung Institute, these therapies have helped greatly. While in the past fewer than half of all people who developed acute respiratory distress syndrome survived, now as many as seven out of ten receiving critical care in a hospital do.
Now, hoping to improve matters further, Rosen and his Scripps Research colleagues are reporting a new molecular mechanism that controls how the lungs are kept dry and under what conditions they permit fluids to enter. The mechanism involves a protein called the S1P3 receptor expressed on the surface of the cells lining the lung's air sacs. When the receptor is activated, the lungs become leaky, causing pulmonary edema.
Because the S1P3 receptor is involved in pulmonary edema, blocking this receptor may be a way to improve the prognosis for people with acute respiratory distress syndrome.
Gas Exchange and What Goes Wrong When Lungs Are Wet
The lung is a remarkable piece of anatomy that enables the exchange of gaseous molecules from the environment with molecules in the bloodstream. Though the lungs are compact enough to fit inside our rib cages, lung tissue is a series of airways and air sacs so elaborate that the air cavities inside the lungs encompass an area about 40 times larger than the surface area of the entire body.
These air cavities play a crucial role for the body because they let oxygen into the bloodstream, where it is picked up by erythrocytes, or red blood cells, and carried to the rest of the tissues throughout the body. The cavities are also where carbon dioxide, a waste product, is removed from the blood stream and expelled from the body.
With each breath you take, air flows in your mouth or through your nasal passageways and down your throat. It goes past the epiglottis, the flap that keeps the food and drink you have consumed from spilling into your lungs. Then, the air flows into the larynx, past your vocal cords, and down the trachea, which splits into the two primary bronchi—one feeding each lung. From there, the air continues to the ends of the bronchi, which bifurcate like thousands of stems branching from a trunk into about 30,000 tiny terminal "bronchioles" in each lung. At the ends of the bronchioles are tiny grape-like clusters of air sacs known as the alveoli. It is in the alveoli that the gas exchange with blood occurs.
The alveoli are elastic cavities lined with a tiny amount of fluid and a molecule called surfactant, which prevents these airways from collapsing in on themselves. Surrounding the alveoli are networks of tiny capillaries that carry oxygen-depleted blood around the outside of the alveoli. When a tiny portion of air reaches the alveoli, gasses are easily dissolved into the fluid, and then exchanged with molecules in the adjacent bloodstream.
One physiology that enables this exchange is the lung epithelium, the specialized layer of cells immediately lining the air sacs. The epithelium is held together via what are known as tight junctions. These tight junctions are made up of proteins that insert through the membranes of adjacent cells and link the cells together so tightly that they prevent salt and other small molecules from passing through the gaps between the cells.
In the airways of the lung, tight junctions are critical because there are only two layers of cells between the air and the blood it is supplying with oxygen—a layer of epithelial cells lining the alveoli and a layer of endothelial cells forming the walls of the blood vessels. The total space between the bloodstream and the air at this interface is only about one five-thousandth of a millimeter. This thinness is essential because oxygen and carbon dioxide molecules have to be able to pass through this space during gas exchange, and the further the molecules have to go, the harder it is for oxygen to reach the blood.
When fluid leaks into the lungs, the distance the gas molecules must travel to reach the blood increases, and this impedes gas exchange. In acute respiratory distress syndrome, pulmonary edema can be so severe that the lungs become heavy and stiff, which is why acute respiratory distress syndrome has also been called "wet lung" or "stiff lung."
Physiologically, pulmonary edema can be caused by conditions other than acute respiratory distress syndrome, for example congestive heart failure, which leads to an increase in pressure in the capillaries surround the lung sacs and a leaking of fluid in the lungs. But in acute respiratory distress syndrome, there is not necessarily too much pressure in the capillaries. So why the fluid-filled lungs?
One reason, Rosen and his colleagues have reported in an upcoming issue of the journal Proceedings of the National Academy of Sciences, may be because of the signaling of a small lipid called sphingosine 1-phosphate (S1P), that is produced at sites of inflammation. The activation of S1P3 receptors in the lung by S1P may be what causes pulmonary edema to arise by causing a breakdown of the epithelial barrier.
Breakdown of the Tight Junctions
The work started as a collaboration between Rosen and his Scripps Research colleague Professor Jerold Chun, both of whom have spent several years studying various lipids and lipid receptor systems in the body including sphingosine 1-phosphate and the lysophosphatidic acid receptors. Sphingosine 1-phosphate is produced or secreted throughout the body, including in the lungs, where it has been found in the lung fluid taken from patients with asthma. The production of sphingosine 1-phosphate is induced as a response to the presence of pro-inflammatory chemicals such as type-1 interferons and tumor necrosis factor, which are both produced during septic shock, one of the exact conditions that lead to edema.
Wanting to know what effect sphingosine 1-phosphate has on edema, the Rosen lab looked at the effect of the lipid on the cells lining the lung and the blood vessels surrounding the lungs. Chun's group had created mutant mice deficient for the S1P3 receptor, which allowed a clean assessment of its role in the lung.
On the blood vessel side, the "endothelial" cells lining the capillaries express a type of protein known as S1P1 receptors. Activation of these S1P1 receptors with the sphingosine 1-phosphate leads to the tightening of junctions between the endothelial cells and the stoppage of potential leakage—the opposite of what happens in edema.
However, the epithelial cells on the lung side express a slightly different type of S1P receptor called the S1P3 receptor protein. Yasuhiro Gon found that when sphingosine 1-phosphate is administered into lung sacs, it activates the S1P3 receptors on the airway side of these epithelial cells and induces pulmonary edema. Significantly, they found that a mouse model that has no receptors of this type is protected against pulmonary edema when exposed to sphingosine 1-phosphate.
Why does the sphingosine 1-phosphate induce lung leakage? To answer this, Rosen and Gon turned to their collaborators Malcolm Wood and William Kiosses in Scripps Research's Core Microscopy facility. They applied fluorescence microscopy to sections of tissue that had been exposed to sphingosine 1-phosphate and showed that the leakage occurs because the activation of S1P3 receptor signaling causes disruptions in the integrity of the tight junctions between epithelial cells. Electron microscopy revealed that certain proteins normally found in the tight junctions had been lost.
These results suggest that a chemical antagonist (something that blocks activation) of the sphingosine 1-phosphate receptors in the lung airways might be protective against pulmonary edema and might lead to a therapy to address acute respiratory distress syndrome.
The article, "S1P3 receptor-induced reorganization of epithelial tight junctions compromises lung barrier integrity and is potentiated by TNF" by Yasuhiro Gon, Malcolm R. Wood, William B. Kiosses, Euijung Jo, M. Germana Sanna, Jerold Chun, and Hugh Rosen will appear in an upcoming issue of the journal Proceedings of the National Academy of Sciences. The article was published June 20, 2005 on the PNAS Early Edition website. See: http://dx.doi.org/10.1073/pnas.0501997102.
This work was supported by grants from the National Institute of Allergy and Infectious Disease, the National Institute of Mental Health, the National Institute of Neurological Disorders and Stroke—all three components of the NIH. Additional support was provided by Kyorin Pharmaceutical Company, Ltd.
For general information on acute respiratory distress syndrome, see: http://www.nhlbi.nih.gov/health/dci/Diseases/Ards/Ards_WhatIs.html
Send comments to: jasonb@scripps.edu
"[Pulmonary edema] is a serious complication that often results in death."
-Hugh Rosen
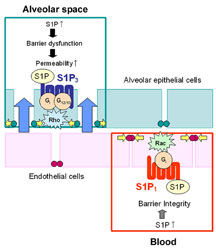
Spatially and mechanistically distinct S1P receptor subtypes have opposing
effects on pulmonary
epithelial and endothelial barriers. Click to Enlarge.
