Mysteries of a Therapy Unveiled
By Jason Socrates Bardi
"Nature
loves to hide."
—Heraclitus
of Ephesus, Fragments, circa 500 B.C.
Several years ago, a blood protein called activated protein C was found
to lower the mortality in patients who acquire severe sepsis. Six months
ago, activated protein C was approved in recombinant form by the Food
and Drug Administration for use in severe sepsis after the protein proved
to be effective at lowering mortality. Today the drug is sold under the
brand name Xigris and is manufactured by Eli Lilly.
Despite its demonstrated efficacy, and despite the fact that scientists
had pondered its beneficial therapeutic effect for a decade, exactly how
activated protein C improved the prognosis for sepsis had remained a mystery.
Now a group of researchers at The Scripps Research Institute (TSRI)
have described how activated protein C works. The group, led by TSRI Associate
Professor Wolfram Ruf, has elucidated the signaling pathway through which
activated protein C works—the receptors on the surface of cells it
binds to and activates—and have published these results in one of
the latest issues of the journal Science.
"For the first time," says Ruf, "we know which players are involved."
The Bacterial Death Knell
Septic shock, also known as sepsis and systemic inflammatory response
syndrome, is a fast-moving, dramatic, and often fatal disease and is a
major problem in U.S. hospitals and hospitals worldwide.
The prognosis for sepsis is dire. According to the National Institutes
of Health, two percent of all hospital admissions suffer from sepsis,
which typically has a 30 percent mortality rate and can be as high as
60 percent. Sepsis is one of the ten leading causes of both infant and
adult mortality in the United States, and directly caused over 30,000
deaths in 1999 alone, according to the Centers for Disease Control and
Prevention (CDC). And the prognosis is especially dire for children.
In a widespread infection, the response of the immune system is triggered
by chemical components of microorganisms, such as endotoxin in certain
bacteria. Endotoxin activates innate immune cells known as monocytes that
induce inflammation at the site of infection. Monocyte/macrophages release
pro-inflammatory cytokines like TNF-a and Interleukin-6
(IL-6), which makes a person feverish. This inflammation is necessary
because without it, the body cannot fight off the bacterial infection.
"This is your first line of defense," says Ruf.
The inflammation that fights the infection can spiral out of control
and lead to septic shock syndrome. One of the signs of severe sepsis is
the activation of coagulation within the vasculature. Platelets disappear
and fibrinogen is consumed. Many different parts of the body can be affected
by this consumptive coagulopathy. Widespread coagulation in the blood
vessels of vital organs leads to blockade of the microcirculation and
whole organs can shut down. Frequently, the vital function of kidneys
and lungs are affected. In patients with sepsis, the levels of inflammatory
cytokines like IL-6 stay high.
"The organ failure is the major problem that results from the inflammation
within the vasculature," says Ruf.
Therapeutic approaches that reduce inflammation proved to make the patients
worse off than they were without treatment because the therapies compromised
their immune response to the bacteria. For many years, the best treatment
has been to administer broad antibiotics to try to quell the infection,
and the rise of antibiotic-resistant bacteria in the last few decades
has promised to exacerbate the problem.
The Protective Protein Pathway
Several years ago, clinical observations led to the idea of using protein
C as a treatment for sepsis. In certain patients, particularly in children
with severe meningococcal sepsis, there was a dramatic decrease in the
level of activated protein C in the blood. Researchers thought that if
this protein was disappearing during severe sepsis, perhaps administering
it to patients would help.
And, indeed, that proved to be the case. Activated protein C fights
inflammation without compromising the body's ability to fight the bacteria
and lowers the mortality due to sepsis. But nobody knew how activated
protein C was mediating anti-inflammatory reactions.
The Ruf laboratory, drawing on several years of work on related areas
of research, figured out the pathway through which activated protein C
works.
The related area that Ruf studies concerns the interaction of proteins
that circulate in the bloodstream and are involved in the blood clotting
cascade with "receptor" proteins, which are displayed on cells on the
inner surface of blood vessels.
The blood clotting cascade is a tightly controlled mechanism designed
primarily to prevent blood loss due to injury, but is also linked to diseases
like cancer and sepsis. During a bacterial infection, when the monocytes
are drawn to a tissue by the presence of endotoxins, they upregulate a
cell surface receptor, tissue factor, which drives blood clotting. Tissue
factor was shown to be an important contributor to the inflammation in
sepsis by animal experiments that were carried out when Ruf was a postdoctoral
fellow in Thomas Edgington's laboratory.
Activation of the coagulation cascade generates thrombin. Thrombin is
a very efficient proteolytic enzyme—it cleaves other proteins at
specific points in their amino acid sequences. One of the proteins it
cleaves is fibrinogen, which makes fibrin, the sticky, clot-forming protein.
Thrombin also cleaves receptor proteins displayed on cell surfaces, which
then "transduce" a signal inside the cell. The first of such a receptor
identified was the thrombin receptor or protease activated receptor 1
(PAR1), a receptor of the broad class of G protein-coupled receptors.
The activation of this receptor on platelets is critical for platelet
activation and hemostasis.
Like all coagulation factors, thrombin circulates as a zymogen—an
inactive precursor. If the body needs to make use of thrombin for the
clotting cascade, for instance, it must first activate the zymogen "prothrombin"
by clipping off the "pro-" part to get thrombin. Obviously, this action
must be tightly controlled by the body to avoid causing blood clots willy-nilly.
One of the ways the body accomplishes this is by requiring prothrombin
to associate with other molecules—called cofactors—before it
can be processed into its mature form. The body uses other enzymes to
control the cofactors and activated protein C is the enzyme that inactivates
the prothrombin cofactor. Activated protein C, thus, in an indirect way
controls blood clotting.
Activated protein C is itself normally present as a zymogen, called
protein C, and it only becomes active when it is cleaved into its active
form. Interestingly, thrombin is the molecule responsible for activating
protein C. Thus the two work together in a feedback loop to balance each
other, thrombin activating the protein C, which deactivates the cofactors
that make thrombin, which reduces the amount of activated protein C, and
so on.
This balance is important for maintaining good health and the feedback
loop that generates activated protein C occurs on the surface of endothelial
cells. Endothelial cells are one of the major cell types of the body,
accounting for about one percent of the total cells in the body. They
line all blood vessels and capillaries and contribute to the structural
integrity of the circulatory system.
Once endothelial cells go, big problems arise—when endothelial
cells suffer widespread damage, organs can fail.
The Mystery Solved
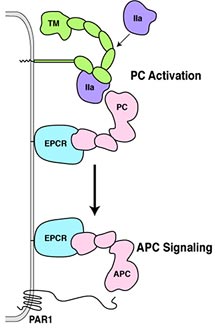
Inactive protein C binds to a specific receptor on the surfaces of these
endothelial cells—called endothelial cell protein C receptor (EPCR).
There, the inactive protein C can be activated by thrombin. To do so,
thrombin needs to bind to another endothelial cell receptor, termed thrombomodulin.
When thrombin is bound to thrombomodulin, thrombin looses all its clotting
function and solely serves to activate the protein C pathway. In sepsis,
the physiological balance between thrombin and activated protein C is
lost, because inflammatory cytokines cause a loss of thrombomodulin from
endothelial cells. Thrombin can no longer activate protein C, and without
activated protein C, the endothelial cells cannot be protected.
Ruf and his colleagues have now shown that ECPR is also required for
activated protein C to trigger a cascade that upregulate genes that prevent
apoptosis, or programmed cell death, and genes that downregulate inflammation
and protect these endothelial cells from damage.
Surprisingly, this cascade involves the thrombin receptor PAR1. PAR1
was the missing link between the activated protein C and its protective
effect on endothelial cells. Clinical trials had established that endothelial
cells can be protected by activated protein C during sepsis, but nobody
knew how.
Ruf and his colleagues demonstrated that activated protein C protected
these endothelial cells through PAR1 signaling by asking whether the genes
that the activated protein C induced could be accounted for by the activation
of the PAR1 receptor.
They ran gene profiling using gene chip microarrays, which allowed them
to look on a genome-wide scale, and they found about 100 genes that are
reproducibly turned on by PAR1. Then they compared those to all the protective
genes that are upregulated by activated protein C, and they found that
they could all be accounted for by the activation of the PAR1 receptor.
Thus, the mystery was solved. Thrombin binds to thrombomodulin on the
surface of endothelial cells and activates the nearby protein C bound
to the EPCR. And activated protein C will then activate the PAR1 receptor.
However, Ruf and his colleagues also found a few genes that were upregulated
by PAR1 signaling, but not by the signaling of activated protein C. Some
of these genes may explain why thrombin has inflammatory effects, while
activated protein C is protective and beneficial. One current direction
of the laboratory is to determine what combinations of gene upregulations
confer protection or cause damage of endothelial cells
The article, "Activation of Endothelial Cell Protease Activated Receptor
1 by the Protein C Pathway" was authored by Matthias Riewald, Ramona J.Petrovan,
Aaron Donner, Barbara M. Mueller, and Wolfram Ruf and appeared in the
June 7, 2002 issue of the journal Science.
Go back to News & Views Index
|