

A Fresh Look at Cystic Fibrosis
By Jason Socrates Bardi
The hundreds of diseases that afflict humankind are named in different ways.
Some are named after an individual, usually the doctor associated with a first descriptive diagnosis of the disease, such as Alois Alzheimer or James Parkinson. Others are named after a place where the disease originated (West Nile virus), where it was first reported (Spanish influenza), or where the first outbreak occurred (Ebola hemorrhagic fever). Others are named after their causes: avian influenza, alcohol poisoning, Guinea worm disease. Still other names derive from a description of the disease's obvious outward symptoms—think smallpox, lockjaw, whooping cough, and yellow fever.
And there are also names that capture the internal pathophysiology of the disease—black lung, for instance, or atherosclerosis, which comes from the Greek athero (meaning gruel or paste) and sclerosis (meaning hardness).
The childhood disease cystic fibrosis is a good example of one of this latter type. Its name refers to two of the hallmarks of the disease—the formation of lung cysts and fibosis, or scar tissue.
Cystic fibrosis is also a good example of a condition that has yielded its molecular secrets to modern biology. We now understand, at least partially, the molecular causes of the disease, and we know, for instance, that cystic fibrosis cases occur due to mutations in a single human gene, called CFTR.
A recent paper in the Journal of Cell Biology by investigators at The Scripps Research Institute presents a fresh view of how CFTR mutations lead to cystic fibrosis.
"This work establishes a new way of looking at the underlying defect that causes the disease," says Scripps Research Professor William Balch, who led the research.
One of The Most Common Genetic Diseases in the U.S.
Cystic fibrosis is one the most common genetic childhood disease in the United States, says Balch, who is a member of the Department of Cell Biology and the Institute for Childhood and Neglected Diseases at Scripps Research. It strikes about one out of every 3,000 Americans born of Caucasian descent, and many children of African-American and Asian descent, but with lower frequency.
Cystic fibrosis occurs when someone inherits a mutant form of a key gene present in the lungs and in many other tissues—the gene CFTR, which is an acronym for cystic fibrosis transmembrane conductance regulator, a protein that was discovered in the late 1980s. About 90 percent of children with cystic fibrosis have the disease as a result of one or more mutations in the CFTR gene, and more than 1000 such mutations have been found to date.
CFTR is an enormous integral membrane protein with about 1,500 amino acids and a complicated structure that spans the cell membrane multiple times in specialized "epithelial" cells. These form in the lining of the lungs, kidneys, and other tissues that produce mucous, sweat, tears, saliva, and other bodily secretions.
CFTR is a chloride channel. When CFTR fails to perform its function, there is an imbalance in the movement of ions and water in and out of the tissue, and the body may lose the ability to regulate the consistency of mucous and other secretions.
The lungs require CFTR chloride channels to function properly as the small air sacs at the termini of the lung's airways need to be bathed continuously in a glycoprotein bath. The CFTR protein normally transports water and ions in and out of the epithelial cells lining these air sacs in order to maintain the consistency of these secretions. In children with cystic fibrosis, defects in the CFTR gene lead to decreased amounts of CFTR proteins on the surface of the cells. Without the CFTR-mediated movement of ions and water, there is not enough water in the lung secretions.
The disease manifests as abnormally thick mucous in the lungs, which leads to obstructed airways, chronic coughing, and bacterial infections in the lungs. Over time, these symptoms can lead to chronic progressive damage to the respiratory system and other organs.
Currently, antibiotics are one of the front-line therapeutics because one common risk for patients is an infection with Pseudomonasaeruginosa, which can lead to an often fatal form of bacterial pneumonia.
Another effective treatment is for parents to beat their children's backs regularly to help them loosen and expel mucous from their lungs.
But currently no FDA-approved treatments can correct the accumulation of abnormally thick mucous in the lungs or the underlying defect that leads to it. Hoping to improve this situation, Balch and his colleagues have engaged in a long course of study aimed at understanding the detailed molecular and cellular mechanisms involved in CFTR and cystic fibrosis.
Identifying the Problem
For years, the prevailing scientific opinion has been that the problem causing cystic fibrosis lies in the body's protein degradation machinery. The basic problem, or so it was thought, was that mutations in the CFTR gene cause the mutant CFTR proteins to be prematurely degraded and thus prevented from reaching the surface of the epithelial cells in the lungs where they are needed.
Studies by scientists in a number of different laboratories showed that the CFTR protein was being degraded in the cells of patients with cystic fibrosis before it ever reached the cells' surfaces. However, the new study by Balch and his colleagues suggests that this degradation is an effect, not a cause, of the underlying problem.
In most kids with cystic fibrosis, says Balch, the CFTR protein gets stuck inside the cells in a cell organelle known as the endoplasmic reticulum—a convoluted membranous sac within the cell where the synthesis of proteins like CFTR and other vital cell functions take place. CFTR's journey through the endoplasmic reticulum begins when the CFTR gene is first transcribed in the nucleus into mRNA, which becomes associated with a ribosome, the molecular machine that translates mRNA into protein, and moves to the endoplasmic reticulum. At the endoplasmic reticulum, the CFTR protein is translated and threaded into the membrane.
From there, the CFTR is packaged into a transport vesicle that goes to a multiple-compartment organelle within the cell known as the Golgi apparatus, where the protein is transferred from compartment to compartment and modified to convert it into its mature 12 transmembrane-domain form. Finally, the mature, functional CFTR is transported to the cell surface where it goes to work.
For the last 15 years or so, Balch and members of his laboratory have been studying the machinery and mechanisms that get CFTR and other cargo out of the endoplasmic reticulum and deliver it to the cell's surface. Now, the group has built on this work by publishing a paper in a recent issue of Journal of Cell Biology that describes an underlying problem caused by the most prevalent mutant form of the CFTR protein.
Mutant CFTR Gets Stuck in Endoplasmic Reticulum
The CFTR mutant that Balch and his colleagues looked at is known as ΔF508, which is the most common CFTR mutation that causes cystic fibrosis, accounting for about 90 percent of cystic fibrosis alleles leading to clinical symptoms.
This mutation does not lead to its premature degradation directly, as had been previously suspected, but rather causes the CFTR protein to become stuck in the endoplasmic reticulum.
This is because in order to make it out of the endoplasmic reticulum, the CFTR must engage a protein complex known as coat complex II (COPII). COPII is responsible for recognizing when the synthesis of CFTR is finished and then grabbing it, packaging it in transport vesicle, and sending it on its way—eventually leading to CFTR's transport to the outside of the cell.
Normally, once the CFTR protein is expressed and folded in the membrane of the ER, it presents its "exit code" to the COPII protein, which then packs it and ships it. But the ΔF508 mutant protein loses its the ability of the exit code to be seen by COPII. It fails to engage the COPII exit machinery and exit the endoplasmic reticulum.
By looking at structures of the domain of CFTR with the exit code and the domain in COPII that recognized the exit code, Balch and his colleagues were able to demonstrate how wild-type CFTR engages COPII and why the ΔF508 mutant protein may not—the loss of the bulky phenylalanine residue at position 508 in the amino acid chain is likely to cause a change in the structure that disrupts this interaction.
CFTR with the ΔF508 mutation is like a passport with a missing page—the picture looks good, the identity is correct, but the exit visa is missing. Without this crucial phenylalanine residue, the CFTR will become stuck in the endoplasmic recticulum and will eventually be degraded as the cell conducts its routine housecleaning.
"For the first time," says Balch, "we understand what it takes to get CFTR protein out of the endoplasmic recticulum."
The next step, he adds, is to identify where in the pathway CFTR's folding is defective and to find ways of targeting the defect to correct folding.
To read the article, "COPII-dependent export of cystic fibrosis transmembrane conductance regulator from the ER uses a di-acidic exit code" by Xiaodong Wang, Jeanne Matteson, Yu An, Bryan Moyer, Jin-San Yoo, Sergei Bannykh, Ian A. Wilson, John R. Riordan, and William E. Balch, see the October 11, 2004 issue of the Journal of Cell Biology (65–74) or go to: http://www.jcb.org/cgi/doi/10.1083/jcb.200401035.
Send comments to: jasonb@scripps.edu

"This work establishes a new way of looking at the underlying defect that causes [cystic fibrosis]," says Professor William Balch. Photo by Kevin Fung.
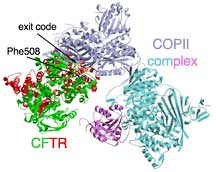
Cystic fibrosis is a good example of a condition that has yielded its molecular secrets to modern biology. Click to enlarge.
